Some Aspects of the Chemistry of Alkynylsilanes
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Porphyrin Carbene Complexes: (5,10,15,20-Tetra-P-Tolylporphyrinato )
Chemistry Publications Chemistry 8-1994 Properties and Molecular Structures of Osmium(ll) Porphyrin Carbene Complexes: (5,10,15,20-Tetra-p-tolylporphyrinato )osmium Di-p-tolylmethylidene and (5,10,15,20-Tetra-p- tolylporphyrinato)osmium (Trimethylsilyl)methylidene Jean-Pierre Djukic Iowa State University Daniel A. Smith Iowa State University Victor G. Young Jr. Iowa State University Follow this and additional works at: http://lib.dr.iastate.edu/chem_pubs L. Keith Woo IowaP Satrate of U ntheiversitCyhe, kmiwoo@istryas Ctaommonte.edu s The ompc lete bibliographic information for this item can be found at http://lib.dr.iastate.edu/ chem_pubs/727. For information on how to cite this item, please visit http://lib.dr.iastate.edu/ howtocite.html. This Article is brought to you for free and open access by the Chemistry at Iowa State University Digital Repository. It has been accepted for inclusion in Chemistry Publications by an authorized administrator of Iowa State University Digital Repository. For more information, please contact [email protected]. Properties and Molecular Structures of Osmium(ll) Porphyrin Carbene Complexes: (5,10,15,20-Tetra-p-tolylporphyrinato )osmium Di-p- tolylmethylidene and (5,10,15,20-Tetra-p-tolylporphyrinato)osmium (Trimethylsilyl)methylidene Abstract The first molecular structures of two (porphyrinato)osmium(II) alkylidene complexes are described. The carbene fragments of (5,10,15,20-tetra-p-tolylporphyrinato)osmium (trimethylsilyl) methylidene (1) and (5,10,15,20-tetra-p-tolylporphyrinato)osmium di-p-tolylmethylidene (2) adopt different conformations in the solid state. With respect to the porphyrin ring nitrogen atoms, a staggered conformation is found for the complex 1 carbene moiety (dos-e = 1. -

Conversion of Carbon Dioxide to Acetylene on a Micro Scale
810 NATURE June 14, 1947 Vol. 159 orbitale of the ethmoid is reduced". In the orang Stainless steel was found to be the most satis and the gibbon a large planum orbitale articulates factory furnace material tried. From mild steel in front with the lacrimal, as in man. The figure relatively large amounts of acetylene were produced we give of the orbital wall in Pleaianthropus shows in blank experiments, and a fused silica envelope a condition almost exactly as in man. fitted with a nickel thimble was found, after it had We are here not at present concerned with the been used with calcium and barium metals, to absorb question of whether man and the Australopithecinre carbon dioxide when hot even when no calcium or have arisen from an early anthropoid, or a pre barium was present. In carrying out the absorption anthropoid, or an Old World monkey or a tarsioid ; of carbon dioxide by barium metal in the stainless but we think the evidence afforded by this new skull steel furnace it was found that when the pressure of Plesianthropus shows that the Australopithecinre at which the gas was admitted was less than about and man are very closely allied, and that these small 10·1 mm. of mercury, the yield of acetylene was brained man-like beings were very nearly human. variable and only about 45 per cent. Good yields R. BROOM were obtained when the carbon dioxide at its full J. T. RoBINSON pressure was admitted to the furnace before raising Transvaal Museum, Pretoria. the temperature above 400° C. -

Synthesis and Characterization of Oligothiophene - Based Fully Π-Conjugated Macrocycles
Synthesis and Characterization of Oligothiophene - based Fully π-Conjugated Macrocycles Dissertation zur Erlangung des Doktorgrades Dr. rer. nat. der Fakultät der Naturwissenschaften der Universität Ulm vorgelegt von Gerda Laura Fuhrmann aus Neustadt (Baia Mare) Rumänien Ulm, 2006 Amtierender Dekan: Prof. Dr. Klaus-Dieter Spindler 1. Gutachter: Prof. Dr. Peter Bäuerle 2. Gutachter: Prof. Dr. Volkhard Austel Tag der Promotionsprüfung: 15.03.2006 This thesis was elaborated and written between November 1999 and February 2006 at the department of Organic Chemistry II, University of Ulm, Germany. für meine Eltern “The chemist does a mysterious thing when he wants to make a molecule. He sees that it has got a ring, so he mixes this and that, and he takes it, and he fiddles around. And at the end of a difficult process, he usually does succeed in synthesizing what he wants.” There’s a Plenty of Room at the Bottom - R. Feynman Ich möchte mich an dieser Stelle bei all denjenigen bedanken, die mich in den zurückliegenden Jahren begleitet und unterstützt haben und damit zum Gelingen dieser Arbeit beigetragen haben. Mein besonderer Dank gilt: - Herrn Prof. Dr. Peter Bäuerle für die stete Unterstützung und Förderung bei der wissenschaftlichen Gestaltung dieser Arbeit, für sein Interesse und persönliches Engagement, sowie für die mir gewähren Freiräume bei der Bearbeitung des Themas - Frau Dr. Pinar Kilickiran für ihre moralische und inspirative Unterstützung, für ihre unermüdliche Hilfe, für die Durchsicht und sprachliche Überarbeitung des Manuskripts und für vieles mehr - Herrn Prof. Dr. V. Austel für die stets anregende wissenschaftliche Diskussionen und seine Bereitschaft diese Arbeit zu begutachten - Herrn Dr. -

Cyanosilylation of Aldehydes Catalyzed by Ag(I)- and Cu(II)-Arylhydrazone Coordination Polymers in Conventional and in Ionic Liquid Media
catalysts Article Cyanosilylation of Aldehydes Catalyzed by Ag(I)- and Cu(II)-Arylhydrazone Coordination Polymers in Conventional and in Ionic Liquid Media Gonçalo A. O. Tiago 1, Kamran T. Mahmudov 1,2,*, M. Fátima C. Guedes da Silva 1,* , Ana P. C. Ribeiro 1,* , Luís C. Branco 3, Fedor I. Zubkov 4 and Armando J. L. Pombeiro 1 1 Centro de Química Estrutural, Instituto Superior Técnico, Universidade de Lisboa, Av. Rovisco Pais, 1049–001 Lisboa, Portugal; [email protected] (G.A.O.T.); [email protected] (A.J.L.P.) 2 Department of Chemistry, Baku State University, Z. Xalilov Str. 23, Az 1148 Baku, Azerbaijan 3 LAQV-REQUINTE, Departamento de Química, Faculdade de Ciências e Tecnologias da Universidade Nova de Lisboa, Quinta da Torre, 2829-516 Caparica, Portugal; [email protected] 4 Organic Chemistry Department, Faculty of Science, Peoples’ Friendship University of Russia (RUDN University), 6 Miklukho-Maklaya St., Moscow 117198, Russian; [email protected] * Correspondence: [email protected] or [email protected] (K.T.M.); [email protected] (M.F.C.G.d.S.); [email protected] (A.P.C.R.) Received: 22 February 2019; Accepted: 15 March 2019; Published: 20 March 2019 0 Abstract: The novel Ag(I) and Cu(II) coordination polymers [Ag(m3-1κO;2:3κO ;4κN-HL)]n·n/2H2O(1) − and [Cu(en)2(m-1κO;2κN-L)]n·nH2O(2) [HL = 2-(2-(1-cyano-2-oxopropylidene)hydrazinyl)benzene sulfonate] were synthesized and characterized by IR and ESI-MS spectroscopies, elemental and single crystal X-ray diffraction analyses. -

Trimethylsilyl Trifluoromethanesulfonate-Mediated Additions to Acetals, Nitrones, and Aminals Chelsea Safran
University of Richmond UR Scholarship Repository Honors Theses Student Research 4-1-2013 Trimethylsilyl trifluoromethanesulfonate-mediated additions to acetals, nitrones, and aminals Chelsea Safran Follow this and additional works at: http://scholarship.richmond.edu/honors-theses Recommended Citation Safran, Chelsea, "Trimethylsilyl trifluoromethanesulfonate-mediated additions to acetals, nitrones, and aminals" (2013). Honors Theses. Paper 71. This Thesis is brought to you for free and open access by the Student Research at UR Scholarship Repository. It has been accepted for inclusion in Honors Theses by an authorized administrator of UR Scholarship Repository. For more information, please contact [email protected]. Trimethylsilyl trifluoromethanesulfonate-mediated additions to acetals, nitrones, and aminals By Chelsea Safran Honors Thesis In Program In Biochemistry and Molecular Biology University of Richmond Richmond, VA Spring 2012 Advisor: Dr. C. Wade Downey This thesis has been accepted as part of the honors requirements in the Program in Biochemistry and Molecular Biology ______________________________ _________________ (advisor signature) (date) ______________________________ _________________ (reader signature) (date) Table of Contents i. Acknowledgements ii ii. Abstract iii iii. Chapter I: Introduction 1-4 iv. Chapter II: Amides 4-15 v. Chapter III: I. Bisthione Synthesis 16-18 II. Reactions with other N,O-acetals 18-22 vi. Chapter IV: I. Additions to Nitrones 22-25 II. Future Work 25 vii. Chapter V: Experimental I. N,O-acetal Formation 25-28 II. Addition to Nitrones 28-29 viii. Chapter VI: References 30 i Acknowledgments I would like to acknowledge my research Dr. Wade Downey for all of his time and dedication to my research for the past two years. -
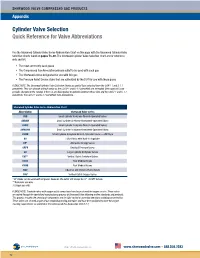
Cylinder Valve Selection Quick Reference for Valve Abbreviations
SHERWOOD VALVE COMPRESSED GAS PRODUCTS Appendix Cylinder Valve Selection Quick Reference for Valve Abbreviations Use the Sherwood Cylinder Valve Series Abbreviation Chart on this page with the Sherwood Cylinder Valve Selection Charts found on pages 73–80. The Sherwood Cylinder Valve Selection Chart are for reference only and list: • The most commonly used gases • The Compressed Gas Association primary outlet to be used with each gas • The Sherwood valves designated for use with this gas • The Pressure Relief Device styles that are authorized by the DOT for use with these gases PLEASE NOTE: The Sherwood Cylinder Valve Selection Charts are partial lists extracted from the CGA V-1 and S-1.1 pamphlets. They can change without notice as the CGA V-1 and S-1.1 pamphlets are amended. Sherwood will issue periodic changes to the catalog. If there is any discrepancy or question between these lists and the CGA V-1 and S-1.1 pamphlets, the CGA V-1 and S-1.1 pamphlets take precedence. Sherwood Cylinder Valve Series Abbreviation Chart Abbreviation Sherwood Valve Series AVB Small Cylinder Acetylene Wrench-Operated Valves AVBHW Small Cylinder Acetylene Handwheel-Operated Valves AVMC Small Cylinder Acetylene Wrench-Operated Valves AVMCHW Small Cylinder Acetylene Handwheel-Operated Valves AVWB Small Cylinder Acetylene Wrench-Operated Valves — WB Style BV Hi/Lo Valves with Built-in Regulator DF* Alternative Energy Valves GRPV Residual Pressure Valves GV Large Cylinder Acetylene Valves GVT** Vertical Outlet Acetylene Valves KVAB Post Medical Valves KVMB Post Medical Valves NGV Industrial and Chrome-Plated Valves YVB† Vertical Outlet Oxygen Valves 1 * DF Valves can be used with all gases; however, the outlet will always be ⁄4"–18 NPT female. -

Reduction of CO2 by Hydrosilanes in the Presence of Formamidinates Of
Reduction of CO2 by Hydrosilanes in the Presence of Formamidinates of Group 13 and 12 Elements Weiheng Huang, Thierry Roisnel, Vincent Dorcet, Clement Orione, Evgueni Kirillov To cite this version: Weiheng Huang, Thierry Roisnel, Vincent Dorcet, Clement Orione, Evgueni Kirillov. Re- duction of CO2 by Hydrosilanes in the Presence of Formamidinates of Group 13 and 12 Elements. Organometallics, American Chemical Society, 2020, 39 (5), pp.698-710. 10.1021/acs.organomet.9b00853. hal-02531302 HAL Id: hal-02531302 https://hal-univ-rennes1.archives-ouvertes.fr/hal-02531302 Submitted on 15 Apr 2020 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Reduction of CO2 by Hydrosilanes in the Presence of Formamidinates of Groups 13 and 12 Elements Weiheng Huang,a Thierry Roisnel,b Vincent Dorcet,b Clement Orione,c and Evgueni Kirillov a,* a Organometallics: Materials and Catalysis laboratories, Univ Rennes, CNRS, ISCR (Institut des Sciences Chimiques de Rennes), UMR 6226, F-35700 Rennes, France b Centre de diffraction X, Univ Rennes, CNRS, ISCR (Institut des Sciences Chimiques de Rennes), UMR 6226, F-35700 Rennes, France c CRMPO, Univ Rennes, CNRS, ISCR (Institut des Sciences Chimiques de Rennes), UMR 6226, F-35700 Rennes, France Graphical Abstract / For the Table of content entrymanuscript Accepted * Correspondence to Evgueni Kirillov ([email protected]); Fax: +33 (0)223236938. -

Development of a Solid-Supported Glaser-Hay Reaction and Utilization in Conjunction with Unnatural Amino Acids
W&M ScholarWorks Dissertations, Theses, and Masters Projects Theses, Dissertations, & Master Projects 2015 Development of a Solid-Supported Glaser-Hay Reaction and Utilization in Conjunction with Unnatural Amino Acids Jessica S. Lampkowski College of William & Mary - Arts & Sciences Follow this and additional works at: https://scholarworks.wm.edu/etd Part of the Organic Chemistry Commons Recommended Citation Lampkowski, Jessica S., "Development of a Solid-Supported Glaser-Hay Reaction and Utilization in Conjunction with Unnatural Amino Acids" (2015). Dissertations, Theses, and Masters Projects. Paper 1539626985. https://dx.doi.org/doi:10.21220/s2-r9jh-9635 This Thesis is brought to you for free and open access by the Theses, Dissertations, & Master Projects at W&M ScholarWorks. It has been accepted for inclusion in Dissertations, Theses, and Masters Projects by an authorized administrator of W&M ScholarWorks. For more information, please contact [email protected]. Development of a Solid-Supported Glaser-Hay Reaction and Utilization in Conjunction with Unnatural Amino Acids Jessica Susan Lampkowski Ida, Michigan B.S. Chemistry, Siena Heights University, 2013 A Thesis presented to the Graduate Faculty of the College of William and Mary in Candidacy for the Degree of Master of Science Chemistry Department The College of William and Mary May, 2015 COMPLIANCE PAGE Research approved by Institutional Biosafety Committee Protocol number: BC-2012-09-13-8113-dyoung01 Date(s) of approval: This protocol will expire on 2015-11-02 APPROVAL PAGE This -

Safety Data Sheet Material Name: SILANE SDS ID: MAT20590
Safety Data Sheet Material Name: SILANE SDS ID: MAT20590 * * * Section 1 - PRODUCT AND COMPANY IDENTIFICATION * * * Manufacturer Information MATHESON TRI-GAS, INC. General Information: 1-800-416-2505 150 Allen Road, Suite 302 Emergency #: 1-800-424-9300 (CHEMTREC) Basking Ridge, NJ 07920 Outside the US: 703-527-3887 (Call collect) Chemical Family hydrides Synonyms MTG MSDS 78; MONOSILANE (SIH4); SILICANE; SILICON HYDRIDE (SIH4); SILICON TETRAHYDRIDE; SILICON HYDRIDE; MONOSILANE; STCC 4920168; UN 2203; H4Si; RTECS: VV1400000 Product Use industrial Usage Restrictions None known. * * * Section 2 - HAZARDS IDENTIFICATION * * * EMERGENCY OVERVIEW Color: colorless Physical Form: gas Odor: unpleasant odor Health Hazards: respiratory tract irritation, skin irritation, eye irritation Physical Hazards: May explode on contact with water. Flammable gas. May cause flash fire. Extremely flammable. May ignite spontaneously on exposure to air. ____________________________________________________________ Page 1 of 10 Issue Date: 03/17/2010 Revision: 1.0201 Print Date: 6/15/2010 Safety Data Sheet Material Name: SILANE SDS ID: MAT20590 POTENTIAL HEALTH EFFECTS Inhalation Short Term: irritation, nausea, headache Long Term: lung damage Skin Short Term: irritation, blisters Long Term: same as effects reported in short term exposure Eye Short Term: irritation, blurred vision Long Term: same as effects reported in short term exposure Ingestion Short Term: frostbite Long Term: no information is available * * * Section 3 - COMPOSITION / INFORMATION ON INGREDIENTS * * * CAS Component Percent 7803-62-5 SILANE 100.0 * * * Section 4 - FIRST AID MEASURES * * * Inhalation If adverse effects occur, remove to uncontaminated area. Give artificial respiration if not breathing. Get immediate medical attention. Skin If frostbite or freezing occur, immediately flush with plenty of lukewarm water (105 -115 F; 41-46 C). -

Hydrosilylation Reactions Catalyzed by Rhenium
molecules ReviewReview HydrosilylationHydrosilylation ReactionsReactions CatalyzedCatalyzed byby RheniumRhenium DuoDuo WeiWei1,2 1,2,, Ruqaya Ruqaya Buhaibeh Buhaibeh 22, ,Yves Yves Canac Canac 22 andand Jean-Baptiste Jean-Baptiste Sortais Sortais 2,3,*2,3, * 11 Univ.University Rennes, Rennes, CNRS, CNRS, ISCR - ISCR-UMR UMR 6226, 6226, F-35000 35000 Rennes, Rennes, France; France; [email protected] [email protected] 22 LCC-CNRS,LCC-CNRS, Université Université dede Toulouse, Toulouse, UPS, UPS, 31400 31400 Toulouse, Toulouse, France; [email protected] (R.B.); [email protected]@lcc-toulouse.fr (Y.C.) (Y.C.) 33 InstitutInstitut Universitaire Universitaire de de France France 1 1 rue rue Descartes, Descartes, 75231 CEDEX Paris 05, Cedex 75231 Paris,05, France France ** Correspondence:Correspondence: [email protected] [email protected] Abstract:Abstract: Hydrosilylation isis anan important process,process, notnot onlyonly inin thethe siliconsilicon industryindustry toto produceproduce siliconsilicon polymers,polymers, butbut alsoalso in finefine chemistry. InIn this review,review, thethe developmentdevelopment ofof rhenium-basedrhenium-based catalystscatalysts forfor thethe hydrosilylationhydrosilylation of of unsaturated unsaturated bonds bonds in in carbonyl-, carbonyl-, cyano-, cyano-, nitro-, nitro-, carboxylic carboxylic acid acid derivatives derivatives and alkenesand alkenes is summarized. is summarized. Mechanisms Mechanisms of rhenium-catalyzed of rhenium-catalyzed -

Kinetic Study of the Selective Hydrogenation of Acetylene Over Supported Palladium Under Tail-End Conditions
catalysts Article Kinetic Study of the Selective Hydrogenation of Acetylene over Supported Palladium under Tail-End Conditions Caroline Urmès 1,2, Jean-Marc Schweitzer 2, Amandine Cabiac 2 and Yves Schuurman 1,* 1 IRCELYON CNRS, UMR 5256, Univ Lyon, Université Claude Bernard Lyon 1, 2 avenue Albert Einstein, 69626 Villeurbanne Cedex, France; [email protected] 2 IFP Energies nouvelles, Etablissement de Lyon, Rond-point de l’échangeur de Solaize, BP3, 69360 Solaize, France; [email protected] (J.-M.S.); [email protected] (A.C.) * Correspondence: [email protected]; Tel.: +33-472445482 Received: 9 January 2019; Accepted: 31 January 2019; Published: 14 February 2019 Abstract: The kinetics of the selective hydrogenation of acetylene in the presence of an excess of ethylene has been studied over a 0.05 wt. % Pd/α-Al2O3 catalyst. The experimental reaction conditions were chosen to operate under intrinsic kinetic conditions, free from heat and mass transfer limitations. The data could be described adequately by a Langmuir–Hinshelwood rate-equation based on a series of sequential hydrogen additions according to the Horiuti–Polanyi mechanism. The mechanism involves a single active site on which both the conversion of acetylene and ethylene take place. Keywords: power-law; Langmuir–Hinshelwood; kinetic modeling; Pd/α-Al2O3 1. Introduction Ethylene is the largest of the basic chemical building blocks with a global market estimated at more than 140 million tons per year with an increasing growth rate. It is used mainly as precursor for polymers production, for instance polyethylene, vinyl chloride, ethylbenzene, or even ethylene oxide synthesis. -

Morphology and Properties of Anti-Corrosion Organosilane Films
UNIVERSITY OF CINCINNATI Date:___________________ I, _________________________________________________________, hereby submit this work as part of the requirements for the degree of: in: It is entitled: This work and its defense approved by: Chair: _______________________________ _______________________________ _______________________________ _______________________________ _______________________________ Morphology and Properties of Anti-Corrosion Organosilane Films A dissertation submitted to the Division of Research and Advanced Studies of the University of Cincinnati in partial fulfillment of the requirements for the degree of DOCTOR OF PHILOSOPHY in the Department of Chemical and Materials Engineering of the College of Engineering 2006 by Guirong Pan B.S., Tongji University, P. R. China 1999 M.S. University of Cincinnati, Ohio, 2003 Committee Chair: Dr. Dale W. Schaefer Abstract Although it is known that certain organosilanes can dramatically improve the corrosion resistance when deposited on metals, the origin of this effect and its dependence on film characteristics are not fully understood. In this work, the morphology and structure of the silane films, as well as their response to water exposure, are studied mainly by neutron reflectivity. Hydrothermal conditioning and solvent swelling are used to challenge the films. The silanes studied include bis-[triethoxysilylpropyl]tetrasulfide (bis-sulfur) and bis- [trimethoxysilylpropyl]amine (bis-amino) as well as mixed silane films. Initial studies were done on films spin-coated on silicon wafer substrates from 1% solutions and cured at 80 °C. Here the focus is the effect of the bridging group on the morphology and water-barrier properties of the films. Subsequent work addresses the same systems deposited on aluminum substrates, films cured at 180 °C and films of larger thickness.