Retinal Genomic Fabric Remodeling After Optic Nerve Injury
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
Peripheral Mononuclear Blood Cells Contribute to The
Genes Nutr (2015) 10:11 DOI 10.1007/s12263-015-0460-8 RESEARCH PAPER Peripheral mononuclear blood cells contribute to the obesity- associated inflammatory state independently of glycemic status: involvement of the novel proinflammatory adipokines chemerin, chitinase-3-like protein 1, lipocalin-2 and osteopontin 1,2,3 1,2,3 1,2,3 Victoria Catala´n • Javier Go´mez-Ambrosi • Amaia Rodrı´guez • 1,2,3 2,4 3,5 2,3,6 Beatriz Ramı´rez • Vı´ctor Valentı´ • Rafael Moncada • Camilo Silva • 2,6 1,2,3,6 Javier Salvador • Gema Fru¨hbeck Received: 24 December 2014 / Accepted: 28 March 2015 / Published online: 14 April 2015 Ó Springer-Verlag Berlin Heidelberg 2015 Abstract Inflammation is a critical contributor to the including chemerin, chitinase-3-like protein 1 (YKL-40), pathogenesis of metabolic disorders with adipose tissue lipocalin-2 (LCN-2) and osteopontin (OPN), and their being crucial in the inflammatory response by releasing circulating concentrations were also determined by ELISA. multiple adipokines with either pro- or anti-inflammatory We show, for the first time, that PBMC gene expression activities with potential functions as metabolic regulators. levels of chemerin (P \ 0.0001), chitinase-3-like protein 1 Peripheral blood mononuclear cells (PBMC) have been (P = 0.010), lipocalin-2 (P \ 0.0001) and osteopontin proposed as representative of the inflammatory status in (P \ 0.0001) were strongly upregulated in obesity inde- obesity. The aim of the present study was to evaluate the pendently of the glycemic state. Circulating concentrations contribution of PBMC to the obesity-associated chronic of these adipokines followed the same trend being sig- inflammation analyzing the expression of novel adipokines. -

Universidade Estadual De Campinas Instituto De Biologia
UNIVERSIDADE ESTADUAL DE CAMPINAS INSTITUTO DE BIOLOGIA VERÔNICA APARECIDA MONTEIRO SAIA CEREDA O PROTEOMA DO CORPO CALOSO DA ESQUIZOFRENIA THE PROTEOME OF THE CORPUS CALLOSUM IN SCHIZOPHRENIA CAMPINAS 2016 1 VERÔNICA APARECIDA MONTEIRO SAIA CEREDA O PROTEOMA DO CORPO CALOSO DA ESQUIZOFRENIA THE PROTEOME OF THE CORPUS CALLOSUM IN SCHIZOPHRENIA Dissertação apresentada ao Instituto de Biologia da Universidade Estadual de Campinas como parte dos requisitos exigidos para a obtenção do Título de Mestra em Biologia Funcional e Molecular na área de concentração de Bioquímica. Dissertation presented to the Institute of Biology of the University of Campinas in partial fulfillment of the requirements for the degree of Master in Functional and Molecular Biology, in the area of Biochemistry. ESTE ARQUIVO DIGITAL CORRESPONDE À VERSÃO FINAL DA DISSERTAÇÃO DEFENDIDA PELA ALUNA VERÔNICA APARECIDA MONTEIRO SAIA CEREDA E ORIENTADA PELO DANIEL MARTINS-DE-SOUZA. Orientador: Daniel Martins-de-Souza CAMPINAS 2016 2 Agência(s) de fomento e nº(s) de processo(s): CNPq, 151787/2F2014-0 Ficha catalográfica Universidade Estadual de Campinas Biblioteca do Instituto de Biologia Mara Janaina de Oliveira - CRB 8/6972 Saia-Cereda, Verônica Aparecida Monteiro, 1988- Sa21p O proteoma do corpo caloso da esquizofrenia / Verônica Aparecida Monteiro Saia Cereda. – Campinas, SP : [s.n.], 2016. Orientador: Daniel Martins de Souza. Dissertação (mestrado) – Universidade Estadual de Campinas, Instituto de Biologia. 1. Esquizofrenia. 2. Espectrometria de massas. 3. Corpo caloso. -

Semaphorin7a Promotes Tumor Growth and Exerts a Pro-Angiogenic Effect in Macrophages of Mammary Tumor-Bearing Mice
ORIGINAL RESEARCH ARTICLE published: 05 February 2014 doi: 10.3389/fphys.2014.00017 Semaphorin7A promotes tumor growth and exerts a pro-angiogenic effect in macrophages of mammary tumor-bearing mice Ramon Garcia-Areas 1, Stephania Libreros 1, Samantha Amat 1, Patricia Keating 2, Roberto Carrio 3, Phillip Robinson 4, Clifford Blieden 5 and Vijaya Iragavarapu-Charyulu 1* 1 Tumor Immunology, Department of Biomedical Sciences, Florida Atlantic University, Boca Raton, FL, USA 2 Immunology, Department of Biological Sciences, Florida Atlantic University, Boca Raton, FL, USA 3 Tumor Immunology, Microbiology and Immunology, University of Miami Miller School of Medicine, Miami, FL, USA 4 Department of Clinical Sciences, Florida Atlantic University, Boca Raton, FL, USA 5 Department of Pathology and Laboratory Medicine, Jackson Memorial Hospital, University of Miami Miller School of Medicine, Miami, FL, USA Edited by: Semaphorins are a large family of molecules involved in axonal guidance during the Michal A. Rahat, Technion - Israel development of the nervous system and have been recently shown to have both Institute for Technology, Israel angiogenic and anti-angiogenic properties. Specifically, semaphorin 7A (SEMA7A) has Reviewed by: been reported to have a chemotactic activity in neurogenesis and to be an immune Andrea Foskett, Texas A&M Health α β Science Center, USA modulator through 1 1integrins. SEMA7A has been shown to promote monocyte Jincai Luo, The University of Tokyo, chemotaxis and induce them to produce proinflammatory mediators. In this study we Japan explored the role of SEMA7A in a murine model of breast cancer. We show that SEMA7A Zahava Vadasz, Bnai-Zion Medical is highly expressed by DA-3 murine mammary tumor cells in comparison to normal Center, Israel mammary cells (EpH4), and that peritoneal elicited macrophages from mammary tumor- *Correspondence: Vijaya Iragavarapu-Charyulu, Tumor bearing mice also express SEMA7A at higher levels compared to those derived from Immunology, Department of normal mice. -

Propranolol-Mediated Attenuation of MMP-9 Excretion in Infants with Hemangiomas
Supplementary Online Content Thaivalappil S, Bauman N, Saieg A, Movius E, Brown KJ, Preciado D. Propranolol-mediated attenuation of MMP-9 excretion in infants with hemangiomas. JAMA Otolaryngol Head Neck Surg. doi:10.1001/jamaoto.2013.4773 eTable. List of All of the Proteins Identified by Proteomics This supplementary material has been provided by the authors to give readers additional information about their work. © 2013 American Medical Association. All rights reserved. Downloaded From: https://jamanetwork.com/ on 10/01/2021 eTable. List of All of the Proteins Identified by Proteomics Protein Name Prop 12 mo/4 Pred 12 mo/4 Δ Prop to Pred mo mo Myeloperoxidase OS=Homo sapiens GN=MPO 26.00 143.00 ‐117.00 Lactotransferrin OS=Homo sapiens GN=LTF 114.00 205.50 ‐91.50 Matrix metalloproteinase‐9 OS=Homo sapiens GN=MMP9 5.00 36.00 ‐31.00 Neutrophil elastase OS=Homo sapiens GN=ELANE 24.00 48.00 ‐24.00 Bleomycin hydrolase OS=Homo sapiens GN=BLMH 3.00 25.00 ‐22.00 CAP7_HUMAN Azurocidin OS=Homo sapiens GN=AZU1 PE=1 SV=3 4.00 26.00 ‐22.00 S10A8_HUMAN Protein S100‐A8 OS=Homo sapiens GN=S100A8 PE=1 14.67 30.50 ‐15.83 SV=1 IL1F9_HUMAN Interleukin‐1 family member 9 OS=Homo sapiens 1.00 15.00 ‐14.00 GN=IL1F9 PE=1 SV=1 MUC5B_HUMAN Mucin‐5B OS=Homo sapiens GN=MUC5B PE=1 SV=3 2.00 14.00 ‐12.00 MUC4_HUMAN Mucin‐4 OS=Homo sapiens GN=MUC4 PE=1 SV=3 1.00 12.00 ‐11.00 HRG_HUMAN Histidine‐rich glycoprotein OS=Homo sapiens GN=HRG 1.00 12.00 ‐11.00 PE=1 SV=1 TKT_HUMAN Transketolase OS=Homo sapiens GN=TKT PE=1 SV=3 17.00 28.00 ‐11.00 CATG_HUMAN Cathepsin G OS=Homo -
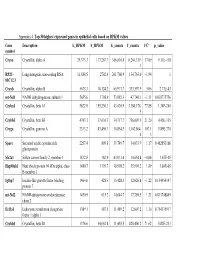
Appendix 4. Top 50 Highest Expressed Genes in Epithelial Cells Based on RPKM Values
Appendix 4. Top 50 highest expressed genes in epithelial cells based on RPKM values Gene Description E_RPKM F_RPKM E_counts F_counts FC* p_value symbol Cryaa Crystallin, alpha A 29,373.3 177,267.7 366,616.4 6,264,319. 17.09 9.11E-118 1 RP23– Long intergenic non-coding RNA 11,888.5 2702.4 261,760.9 134,763.0 −1.94 1 81C12.3 Cryab Crystallin, alpha B 5673.3 10,124.2 65,971.7 333,597.9 5.06 2.71E-43 mt-Nd1 NADH dehydrogenase, subunit 1 5655.6 1798.9 53,082.3 47,748.1 −1.11 0.838775756 Cryba1 Crystallin, beta A1 5622.0 155,230.3 43,420.9 3,380,176. 77.85 1.34E-240 5 Crybb3 Crystallin, beta B3 4743.1 37,636.3 34,717.7 736,007.9 21.20 4.45E-135 Cryga Crystallin, gamma A 2333.2 83,496.3 10,854.5 1,162,864. 107.1 5.89E-270 6 3 Sparc Secreted acidic cysteine rich 2257.4 809.8 39,749.7 34,033.9 −1.17 0.462853166 glycoprotein Slc2a1 Solute carrier family 2, member 1 1832.8 162.9 43,031.4 10,654.8 −4.04 1.67E-05 Hsp90ab1 Heat shock protein 90 kDa alpha, class 1480.7 1139.7 18,998.2 35,901.2 1.89 3.84E-05 B member 1 Igfbp7 Insulin-like growth factor binding 1464.6 428.3 15,428.3 12,626.8 −1.22 0.154954147 protein 7 mt-Nd2 NADH-ubiquinone oxidoreductase 1450.9 615.2 14,644.7 17,789.5 1.21 0.833748849 chain 2 Eef1a1 Eukaryotic translation elongation 1389.1 587.5 11,489.2 12,607.2 1.10 0.754135917 factor 1 alpha 1 Crybb1 Crystallin, beta B1 1376.6 34,662.8 11,455.5 820,406.2 71.62 5.82E-233 Htra3 HtrA serine peptidase 3 1338.6 162.0 23,197.6 6433.9 −3.61 3.93E-05 Gnb2l1 Guanine nucleotide-binding protein 1293.3 670.1 14,495.1 21,652.1 1.49 0.001685952 -

CRYGB Rabbit Pab
Leader in Biomolecular Solutions for Life Science CRYGB Rabbit pAb Catalog No.: A14569 Basic Information Background Catalog No. Crystallins are separated into two classes: taxon-specific, or enzyme, and ubiquitous. A14569 The latter class constitutes the major proteins of vertebrate eye lens and maintains the transparency and refractive index of the lens. Since lens central fiber cells lose their Observed MW nuclei during development, these crystallins are made and then retained throughout 21kDa life, making them extremely stable proteins. Mammalian lens crystallins are divided into alpha, beta, and gamma families; beta and gamma crystallins are also considered as a Calculated MW superfamily. Alpha and beta families are further divided into acidic and basic groups. 20kDa Seven protein regions exist in crystallins: four homologous motifs, a connecting peptide, and N- and C-terminal extensions. Gamma-crystallins are a homogeneous group of Category highly symmetrical, monomeric proteins typically lacking connecting peptides and terminal extensions. They are differentially regulated after early development. Four Primary antibody gamma-crystallin genes (gamma-A through gamma-D) and three pseudogenes (gamma-E, gamma-F, gamma-G) are tandemly organized in a genomic segment as a Applications gene cluster. Whether due to aging or mutations in specific genes, gamma-crystallins WB have been involved in cataract formation. Cross-Reactivity Mouse, Rat Recommended Dilutions Immunogen Information WB 1:500 - 1:2000 Gene ID Swiss Prot 1419 P07316 Immunogen Recombinant fusion protein containing a sequence corresponding to amino acids 80-135 of human CRYGB (NP_005201.2). Synonyms CRYGB;CRYG2;CTRCT39 Contact Product Information www.abclonal.com Source Isotype Purification Rabbit IgG Affinity purification Storage Store at -20℃. -

Defining Functional Interactions During Biogenesis of Epithelial Junctions
ARTICLE Received 11 Dec 2015 | Accepted 13 Oct 2016 | Published 6 Dec 2016 | Updated 5 Jan 2017 DOI: 10.1038/ncomms13542 OPEN Defining functional interactions during biogenesis of epithelial junctions J.C. Erasmus1,*, S. Bruche1,*,w, L. Pizarro1,2,*, N. Maimari1,3,*, T. Poggioli1,w, C. Tomlinson4,J.Lees5, I. Zalivina1,w, A. Wheeler1,w, A. Alberts6, A. Russo2 & V.M.M. Braga1 In spite of extensive recent progress, a comprehensive understanding of how actin cytoskeleton remodelling supports stable junctions remains to be established. Here we design a platform that integrates actin functions with optimized phenotypic clustering and identify new cytoskeletal proteins, their functional hierarchy and pathways that modulate E-cadherin adhesion. Depletion of EEF1A, an actin bundling protein, increases E-cadherin levels at junctions without a corresponding reinforcement of cell–cell contacts. This unexpected result reflects a more dynamic and mobile junctional actin in EEF1A-depleted cells. A partner for EEF1A in cadherin contact maintenance is the formin DIAPH2, which interacts with EEF1A. In contrast, depletion of either the endocytic regulator TRIP10 or the Rho GTPase activator VAV2 reduces E-cadherin levels at junctions. TRIP10 binds to and requires VAV2 function for its junctional localization. Overall, we present new conceptual insights on junction stabilization, which integrate known and novel pathways with impact for epithelial morphogenesis, homeostasis and diseases. 1 National Heart and Lung Institute, Faculty of Medicine, Imperial College London, London SW7 2AZ, UK. 2 Computing Department, Imperial College London, London SW7 2AZ, UK. 3 Bioengineering Department, Faculty of Engineering, Imperial College London, London SW7 2AZ, UK. 4 Department of Surgery & Cancer, Faculty of Medicine, Imperial College London, London SW7 2AZ, UK. -

Bioinformatic Analysis of Structure and Function of LIM Domains of Human Zyxin Family Proteins
International Journal of Molecular Sciences Article Bioinformatic Analysis of Structure and Function of LIM Domains of Human Zyxin Family Proteins M. Quadir Siddiqui 1,† , Maulik D. Badmalia 1,† and Trushar R. Patel 1,2,3,* 1 Alberta RNA Research and Training Institute, Department of Chemistry and Biochemistry, University of Lethbridge, 4401 University Drive, Lethbridge, AB T1K 3M4, Canada; [email protected] (M.Q.S.); [email protected] (M.D.B.) 2 Department of Microbiology, Immunology and Infectious Disease, Cumming School of Medicine, University of Calgary, 3330 Hospital Drive, Calgary, AB T2N 4N1, Canada 3 Li Ka Shing Institute of Virology, University of Alberta, Edmonton, AB T6G 2E1, Canada * Correspondence: [email protected] † These authors contributed equally to the work. Abstract: Members of the human Zyxin family are LIM domain-containing proteins that perform critical cellular functions and are indispensable for cellular integrity. Despite their importance, not much is known about their structure, functions, interactions and dynamics. To provide insights into these, we used a set of in-silico tools and databases and analyzed their amino acid sequence, phylogeny, post-translational modifications, structure-dynamics, molecular interactions, and func- tions. Our analysis revealed that zyxin members are ohnologs. Presence of a conserved nuclear export signal composed of LxxLxL/LxxxLxL consensus sequence, as well as a possible nuclear localization signal, suggesting that Zyxin family members may have nuclear and cytoplasmic roles. The molecular modeling and structural analysis indicated that Zyxin family LIM domains share Citation: Siddiqui, M.Q.; Badmalia, similarities with transcriptional regulators and have positively charged electrostatic patches, which M.D.; Patel, T.R. -

Identification of Genes Concordantly Expressed with Atoh1 During Inner Ear Development
Original Article doi: 10.5115/acb.2011.44.1.69 pISSN 2093-3665 eISSN 2093-3673 Identification of genes concordantly expressed with Atoh1 during inner ear development Heejei Yoon, Dong Jin Lee, Myoung Hee Kim, Jinwoong Bok Department of Anatomy, Brain Korea 21 Project for Medical Science, College of Medicine, Yonsei University, Seoul, Korea Abstract: The inner ear is composed of a cochlear duct and five vestibular organs in which mechanosensory hair cells play critical roles in receiving and relaying sound and balance signals to the brain. To identify novel genes associated with hair cell differentiation or function, we analyzed an archived gene expression dataset from embryonic mouse inner ear tissues. Since atonal homolog 1a (Atoh1) is a well known factor required for hair cell differentiation, we searched for genes expressed in a similar pattern with Atoh1 during inner ear development. The list from our analysis includes many genes previously reported to be involved in hair cell differentiation such as Myo6, Tecta, Myo7a, Cdh23, Atp6v1b1, and Gfi1. In addition, we identified many other genes that have not been associated with hair cell differentiation, including Tekt2, Spag6, Smpx, Lmod1, Myh7b, Kif9, Ttyh1, Scn11a and Cnga2. We examined expression patterns of some of the newly identified genes using real-time polymerase chain reaction and in situ hybridization. For example, Smpx and Tekt2, which are regulators for cytoskeletal dynamics, were shown specifically expressed in the hair cells, suggesting a possible role in hair cell differentiation or function. Here, by re- analyzing archived genetic profiling data, we identified a list of novel genes possibly involved in hair cell differentiation. -

Variants in PAX6, PITX3 and HSF4 Causing Autosomal Dominant Congenital Cataracts ✉ ✉ Vanita Berry 1,2 , Alex Ionides2, Nikolas Pontikos 1,2, Anthony T
www.nature.com/eye ARTICLE OPEN Variants in PAX6, PITX3 and HSF4 causing autosomal dominant congenital cataracts ✉ ✉ Vanita Berry 1,2 , Alex Ionides2, Nikolas Pontikos 1,2, Anthony T. Moore2, Roy A. Quinlan3 and Michel Michaelides 1,2 © Crown 2021 BACKGROUND: Lens development is orchestrated by transcription factors. Disease-causing variants in transcription factors and their developmental target genes are associated with congenital cataracts and other eye anomalies. METHODS: Using whole exome sequencing, we identified disease-causing variants in two large British families and one isolated case with autosomal dominant congenital cataract. Bioinformatics analysis confirmed these disease-causing mutations as rare or novel variants, with a moderate to damaging pathogenicity score, with testing for segregation within the families using direct Sanger sequencing. RESULTS: Family A had a missense variant (c.184 G>A; p.V62M) in PAX6 and affected individuals presented with nuclear cataract. Family B had a frameshift variant (c.470–477dup; p.A160R*) in PITX3 that was also associated with nuclear cataract. A recurrent missense variant in HSF4 (c.341 T>C; p.L114P) was associated with congenital cataract in a single isolated case. CONCLUSIONS: We have therefore identified novel variants in PAX6 and PITX3 that cause autosomal dominant congenital cataract. Eye; https://doi.org/10.1038/s41433-021-01711-x INTRODUCTION consistent with early developmental effects as would be Cataract the opacification of the eye lens is the most common, but anticipated for PAX6 and PITX3 transcription factors. Recently, treatable cause of blindness in the world (https://www.who.int/ we have found two novel mutations in the transcription factors publications-detail/world-report-on-vision). -

Absence of NEFL in Patient-Specific Neurons in Early-Onset Charcot-Marie-Tooth Neuropathy Markus T
ARTICLE OPEN ACCESS Absence of NEFL in patient-specific neurons in early-onset Charcot-Marie-Tooth neuropathy Markus T. Sainio, MSc, Emil Ylikallio, MD, PhD, Laura M¨aenp¨a¨a, MSc, Jenni Lahtela, PhD, Pirkko Mattila, PhD, Correspondence Mari Auranen, MD, PhD, Johanna Palmio, MD, PhD, and Henna Tyynismaa, PhD Dr. Tyynismaa [email protected] Neurol Genet 2018;4:e244. doi:10.1212/NXG.0000000000000244 Abstract Objective We used patient-specific neuronal cultures to characterize the molecular genetic mechanism of recessive nonsense mutations in neurofilament light (NEFL) underlying early-onset Charcot- Marie-Tooth (CMT) disease. Methods Motor neurons were differentiated from induced pluripotent stem cells of a patient with early- onset CMT carrying a novel homozygous nonsense mutation in NEFL. Quantitative PCR, protein analytics, immunocytochemistry, electron microscopy, and single-cell transcriptomics were used to investigate patient and control neurons. Results We show that the recessive nonsense mutation causes a nearly total loss of NEFL messenger RNA (mRNA), leading to the complete absence of NEFL protein in patient’s cultured neurons. Yet the cultured neurons were able to differentiate and form neuronal networks and neuro- filaments. Single-neuron gene expression fingerprinting pinpointed NEFL as the most down- regulated gene in the patient neurons and provided data of intermediate filament transcript abundancy and dynamics in cultured neurons. Blocking of nonsense-mediated decay partially rescued the loss of NEFL mRNA. Conclusions The strict neuronal specificity of neurofilament has hindered the mechanistic studies of re- cessive NEFL nonsense mutations. Here, we show that such mutation leads to the absence of NEFL, causing childhood-onset neuropathy through a loss-of-function mechanism. -

Experimental Validation of the Regulated Expression of Large Numbers of Non-Coding Rnas from the Mouse Genome
Downloaded from genome.cshlp.org on September 30, 2021 - Published by Cold Spring Harbor Laboratory Press Article Experimental validation of the regulated expression of large numbers of non-coding RNAs from the mouse genome Timothy Ravasi,1,4,5 Harukazu Suzuki,2,4 Ken C. Pang,1,3,4 Shintaro Katayama,2,4 Masaaki Furuno,2,4,6 Rie Okunishi,2 Shiro Fukuda,2 Kelin Ru,1 Martin C. Frith,1,2 M. Milena Gongora,1 Sean M. Grimmond,1 David A. Hume,1 Yoshihide Hayashizaki,2 and John S. Mattick1,7 1ARC Special Research Centre for Functional and Applied Genomics, Institute for Molecular Bioscience, University of Queensland, Brisbane QLD 4072, Australia; 2Laboratory for Genome Exploration Research Group, RIKEN Genomic Science Center, RIKEN Yokohama Institute, Suehiro-cho, Tsurumi-ku, Yokohama, Kanagawa, 230-0045, Japan; 3T Cell Laboratory, Ludwig Institute for Cancer Research, Austin & Repatriation Medical Centre, Heidelberg VIC 3084, Australia Recent large-scale analyses of mainly full-length cDNA libraries generated from a variety of mouse tissues indicated that almost half of all representative cloned sequences did not contain an apparent protein-coding sequence, and were putatively derived from non-protein-coding RNA (ncRNA) genes. However, many of these clones were singletons and the majority were unspliced, raising the possibility that they may be derived from genomic DNA or unprocessed pre-mRNA contamination during library construction, or alternatively represent nonspecific “transcriptional noise.” Here we show, using reverse transcriptase-dependent PCR, microarray, and Northern blot analyses, that many of these clones were derived from genuine transcripts of unknown function whose expression appears to be regulated.