Molecular Genetics of Infantile-Onset Retinal Dystrophies
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Genetic Analysis of Retinopathy in Type 1 Diabetes
Genetic Analysis of Retinopathy in Type 1 Diabetes by Sayed Mohsen Hosseini A thesis submitted in conformity with the requirements for the degree of Doctor of Philosophy Institute of Medical Science University of Toronto © Copyright by S. Mohsen Hosseini 2014 Genetic Analysis of Retinopathy in Type 1 Diabetes Sayed Mohsen Hosseini Doctor of Philosophy Institute of Medical Science University of Toronto 2014 Abstract Diabetic retinopathy (DR) is a leading cause of blindness worldwide. Several lines of evidence suggest a genetic contribution to the risk of DR; however, no genetic variant has shown convincing association with DR in genome-wide association studies (GWAS). To identify common polymorphisms associated with DR, meta-GWAS were performed in three type 1 diabetes cohorts of White subjects: Diabetes Complications and Control Trial (DCCT, n=1304), Wisconsin Epidemiologic Study of Diabetic Retinopathy (WESDR, n=603) and Renin-Angiotensin System Study (RASS, n=239). Severe (SDR) and mild (MDR) retinopathy outcomes were defined based on repeated fundus photographs in each study graded for retinopathy severity on the Early Treatment Diabetic Retinopathy Study (ETDRS) scale. Multivariable models accounted for glycemia (measured by A1C), diabetes duration and other relevant covariates in the association analyses of additive genotypes with SDR and MDR. Fixed-effects meta- analysis was used to combine the results of GWAS performed separately in WESDR, ii RASS and subgroups of DCCT, defined by cohort and treatment group. Top association signals were prioritized for replication, based on previous supporting knowledge from the literature, followed by replication in three independent white T1D studies: Genesis-GeneDiab (n=502), Steno (n=936) and FinnDiane (n=2194). -

Masqueraders of Age-Related Macular Degeneration
COVER STORY Masqueraders of Age-related Macular Degeneration A number of inherited retinal diseases phenocopy AMD. BY RONY GELMAN, MD, MS; AND STEPHEN H. TSANG, MD, PHD ge-related macular degeneration (AMD) is a leading cause of central visual loss among the elderly population in the developed world. The Currently, there are no published A non-neovascular form is characterized by mac- guidelines to prognosticate ular drusen and other abnormalities of the retinal pigment epithelium (RPE) such as geographic atrophy (GA) and Stargardt macular degeneration. hyperpigmented areas in the macula. The neovascular form is heralded by choroidal neovascularization (CNV), with subsequent development of disciform scarring. ABCA4 defect heterozygote carrier may be as high as This article reviews the pathologic and diagnostic char- one in 20.11,12 An estimated 600 disease-causing muta- acteristics of inherited diseases that may masquerade as tions in the ABCA4 gene exist, of which the three most AMD. The review is organized by the following patterns common mutations account for less than 10% of the of inheritance: autosomal recessive (Stargardt disease and disease phenotypes.13 cone dystrophy); autosomal dominant (cone dystrophy, The underlying pathology of disease in STGD involves adult vitelliform dystrophy, pattern dystrophy, North accumulation of lipofuscin in the RPE through a process Carolina macular dystrophy, Doyne honeycomb dystro- of disc shedding and phagocytosis.14,15 Lipofuscin is toxic phy, and Sorsby macular dystrophy); X-linked (X-linked to the RPE; furthermore, A2E, a component of lipofuscin, retinoschisis); and mitochondrial (maternally inherited causes inhibition of 11-cis retinal regeneration16 and diabetes and deafness). complement activation. -
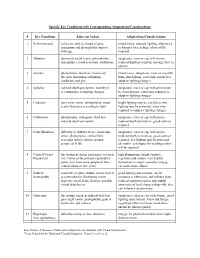
Specific Eye Conditions with Corresponding Adaptations/Considerations
Specific Eye Conditions with Corresponding Adaptations/Considerations # Eye Condition Effect on Vision Adaptations/Considerations 1 Achromotopsia colors are seen as shades of grey, tinted lenses, reduced lighting, alternative nystagmus and photophobia improve techniques for teaching colors will be with age required 2 Albinism decreased visual acuity, photophobia, sunglasses, visor or cap with a brim, nystagmus, central scotomas, strabismus reduced depth perception, moving close to objects 3 Aniridia photophobia, field loss, vision may tinted lenses, sunglasses, visor or cap with fluctuate depending on lighting brim, dim lighting, extra time required to conditions and glare adapt to lighting changes 4 Aphakia reduced depth perception, inability to sunglasses, visor or cap with a brim may accommodate to lighting changes be worn indoors, extra time required to adapt to lighting changes 5 Cataracts poor color vision, photophobia, visual bright lighting may be a problem, low acuity fluctuates according to light lighting may be preferred, extra time required to adapt to lighting changes 6 Colobomas photophobia, nystagmus, field loss, sunglasses, visor or cap with a brim, reduced depth perception reduced depth perception, good contrast required 7 Color Blindness difficulty or inability to see colors and sunglasses, visor or cap with a brim, detail, photophobia, central field reduced depth perception, good contrast scotomas (spotty vision), normal required, low lighting may be preferred, peripheral fields alternative techniques for teaching colors -

Multipurpose Conical Orbital Implant in Evisceration
Ophthalmic Plastic and Reconstructive Surgery Vol. 21, No. 5, pp 376–378 ©2005 The American Society of Ophthalmic Plastic and Reconstructive Surgery, Inc. Multipurpose Conical Orbital Implant in Evisceration Harry Marshak, M.D., and Steven C. Dresner, M.D. Doheny Eye Institute, Keck School of Medicine, University of Southern California, Los Angeles, California, U.S.A. Purpose: To evaluate the safety and efficacy of the porous polyethylene multipurpose conical orbital implant for use in evisceration. Methods: A retrospective review of 31 eyes that underwent evisceration and received the multipurpose conical orbital implant. The orbits were evaluated at 1 week, 1 month, and 6 months after final prosthetic fitting for implant exposure, superior sulcus deformity, and prosthetic motility. Results: There were no cases of extrusion, migration, or infection. All patients had a good cosmetic result after final prosthetic fitting. Prosthetic motility was good in all patients. Exposure developed in one eye (3%) and a superior sulcus deformity developed in one eye (3%). Conclusions: Placement of an multipurpose conical orbital implant in conjunction with evisceration is a safe and effective treatment for blind painful eye that achieves good motility and a good cosmetic result. visceration has proved to be effective for the treat- forms anteriorly to the sclera to be closed over it, without Ement of blind painful eye from phthisis bulbi or crowding the fornices, and extends posteriorly through endophthalmitis. By retaining the sclera in its anatomic the posterior sclerotomies, providing needed volume to natural position, evisceration has the advantage of allow- the posterior orbit. ing the insertions of the extraocular muscles to remain intact, promoting better motility. -

Ametropia and Emmetropization in CNGB3 Achromatopsia
Retina Ametropia and Emmetropization in CNGB3 Achromatopsia Mette Kjøbæk Gundestrup Andersen1 and Line Kessel1,2 1Department of Ophthalmology, Copenhagen University Hospital, Rigshospitalet-Glostrup, Glostrup, Denmark 2Department of Clinical Medicine, University of Copenhagen, Copenhagen, Denmark Correspondence: Mette K.G. PURPOSE. Emmetropization is the process of adjusting ocular growth to the focal plane Andersen, Department of in order to achieve a clear image. Chromatic light may be involved as a cue to guide Ophthalmology, Copenhagen this process. Achromats are color blind and lack normal cone function; they are often University Hospital, described as being hyperopic, indicating a failure to emmetropize. We aim to describe Rigshospitalet-Glostrup, Valdemar the refraction and refractive development in a population of genetically characterized Hansens Vej 1-23, 2600 Glostrup, Denmark; achromats. [email protected]. METHODS. Refractive error data were collected retrospectively from 28 medical records CNGB3 Received: August 23, 2020 of c.1148delC homozygous achromats. The distribution of spherical equivalent Accepted: January 18, 2021 refractive error (SER) and spherical error was analyzed in adults. The refractive develop- Published: February 9, 2021 ment in children was analyzed by documenting astigmatic refractive error and calculating Citation: Andersen MKG, Kessel L. median SER in 1-year age groups and by analyzing the individual development when Ametropia and emmetropization in possible. CNGB3 Invest achromatopsia. RESULTS. The distribution of SER and spherical error resembled a Gaussian distribution, Ophthalmol Vis Sci. 2021;62(2):10. indicating that emmetropization was disturbed in achromats, but we found indication of https://doi.org/10.1167/iovs.62.2.10 some decrease in SER during the first years of childhood. -

Blue Cone Monochromacy: Visual Function and Efficacy Outcome Measures for Clinical Trials
RESEARCH ARTICLE Blue Cone Monochromacy: Visual Function and Efficacy Outcome Measures for Clinical Trials Xunda Luo1☯‡, Artur V. Cideciyan1☯‡*, Alessandro Iannaccone2, Alejandro J. Roman1, Lauren C. Ditta2, Barbara J. Jennings2, Svetlana A. Yatsenko3, Rebecca Sheplock1, Alexander Sumaroka1, Malgorzata Swider1, Sharon B. Schwartz1, Bernd Wissinger4, Susanne Kohl4, Samuel G. Jacobson1* 1 Scheie Eye Institute, Department of Ophthalmology, Perelman School of Medicine, University of Pennsylvania, Philadelphia, Pennsylvania, United States of America, 2 Hamilton Eye Institute, Department of Ophthalmology, University of Tennessee Health Science Center, Memphis, Tennessee, United States of America, 3 Pittsburgh Cytogenetics Laboratory, Center for Medical Genetics and Genomics, University of Pittsburgh School of Medicine, Pittsburgh, Pennsylvania, United States of America, 4 Molecular Genetics Laboratory, Institute for Ophthalmic Research, Centre for Ophthalmology, University of Tuebingen, Tuebingen, Germany ☯ These authors contributed equally to this work. ‡ OPEN ACCESS These authors are joint first authors on this work. * [email protected] (SGJ); [email protected] (AVC) Citation: Luo X, Cideciyan AV, Iannaccone A, Roman AJ, Ditta LC, Jennings BJ, et al. (2015) Blue Cone Monochromacy: Visual Function and Efficacy Abstract Outcome Measures for Clinical Trials. PLoS ONE 10(4): e0125700. doi:10.1371/journal.pone.0125700 Academic Editor: Dror Sharon, Hadassah-Hebrew University Medical Center, ISRAEL Background Blue Cone Monochromacy (BCM) is an X-linked retinopathy caused by mutations in the Received: December 29, 2014 OPN1LW / OPN1MW gene cluster, encoding long (L)- and middle (M)-wavelength sensitive Accepted: March 21, 2015 cone opsins. Recent evidence shows sufficient structural integrity of cone photoreceptors in Published: April 24, 2015 BCM to warrant consideration of a gene therapy approach to the disease. -

A Patient & Parent Guide to Strabismus Surgery
A Patient & Parent Guide to Strabismus Surgery By George R. Beauchamp, M.D. Paul R. Mitchell, M.D. Table of Contents: Part I: Background Information 1. Basic Anatomy and Functions of the Extra-ocular Muscles 2. What is Strabismus? 3. What Causes Strabismus? 4. What are the Signs and Symptoms of Strabismus? 5. Why is Strabismus Surgery Performed? Part II: Making a Decision 6. What are the Options in Strabismus Treatment? 7. The Preoperative Consultation 8. Choosing Your Surgeon 9. Risks, Benefits, Limitations and Alternatives to Surgery 10. How is Strabismus Surgery Performed? 11. Timing of Surgery Part III: What to Expect Around the Time of Surgery 12. Before Surgery 13. During Surgery 14. After Surgery 15. What are the Potential Complications? 16. Myths About Strabismus Surgery Part IV: Additional Matters to Consider 17. About Children and Strabismus Surgery 18. About Adults and Strabismus Surgery 19. Why if May be Important to a Person to Have Strabismus Surgery (and How Much) Part V: A Parent’s Perspective on Strabismus Surgery 20. My Son’s Diagnosis and Treatment 21. Growing Up with Strabismus 22. Increasing Signs that Surgery Was Needed 23. Making the Decision to Proceed with Surgery 24. Explaining Eye Surgery to My Son 25. After Surgery Appendix Part I: Background Information Chapter 1: Basic Anatomy and Actions of the Extra-ocular Muscles The muscles that move the eye are called the extra-ocular muscles. There are six of them on each eye. They work together in pairs—complementary (or yoke) muscles pulling the eyes in the same direction(s), and opposites (or antagonists) pulling the eyes in opposite directions. -

Genetic Defects of CHM and Visual Acuity Outcome in 24 Choroideremia
www.nature.com/scientificreports OPEN Genetic defects of CHM and visual acuity outcome in 24 choroideremia patients from 16 Japanese families Takaaki Hayashi1,2*, Shuhei Kameya3, Kei Mizobuchi2, Daiki Kubota3, Sachiko Kikuchi3, Kazutoshi Yoshitake4, Atsushi Mizota5, Akira Murakami6, Takeshi Iwata4 & Tadashi Nakano2 Choroideremia (CHM) is an incurable progressive chorioretinal dystrophy. Little is known about the natural disease course of visual acuity in the Japanese population. We aimed to investigate the genetic spectrum of the CHM gene and visual acuity outcomes in 24 CHM patients from 16 Japanese families. We measured decimal best-corrected visual acuity (BCVA) at presentation and follow-up, converted to logMAR units for statistical analysis. Sanger and/or whole-exome sequencing were performed to identify pathogenic CHM variants/deletions. The median age at presentation was 37.0 years (range, 5–76 years). The mean follow-up interval was 8.2 years. BCVA of the better-seeing eye at presentation was signifcantly worsened with increasing age (r = 0.515, p < 0.01), with a high rate of BCVA decline in patients > 40 years old. A Kaplan–Meier survival curve suggested that a BCVA of Snellen equivalent 20/40 at follow-up remains until the ffties. Fourteen pathogenic variants, 6 of which were novel [c.49 + 5G > A, c.116 + 5G > A, p.(Gly176Glu, Glu177Ter), p.Tyr531Ter, an exon 2 deletion, and a 5.0-Mb deletion], were identifed in 15 families. No variant was found in one family only. Our BCVA outcome data are useful for predicting visual prognosis and determining the timing of intervention in Japanese patients with CHM variants. -

Refractive Error Changes in Cortical, Nuclear and Posterior Subcapsular Cataracts
1524 Refractive error changes in cortical, nuclear and posterior subcapsular cataracts. D.B. Elliott & K. Pesudovs . Department of Optometry, University of Bradford, Bradford, United Kingdom.. Purpose • To determine the effect of the three main morphological types of age-related cataract on refractive error. Background • The increase in myopia in some patients with age-related nuclear cataract is well known (Brown & Hill, 1987). This change accounts for the “second sight of the elderly” in which the myopic shift provides normal reading ability without the need for spectacles. Distance vision, however, worsens. • Planter (1981) suggested that cortical opacity can induce hyperopic shifts. However, this claim has not been repeated since and no firm evidence is available. • Review papers suggest that cortical opacity can induce astigmatic changes (Brown, 1993). However, these reports were based on clinical impression with no supporting data. • . The prevalence of uncorrected refractive error in the elderly population is high (Wormald et al., 1992). It is likely that a reasonable proportion of these patients have refractive error changes induced by cataract. • It has been suggested that a delay in operating on age-related cataract of 10 years could reduce the number of patients requiring cataract surgery (Kupfer, 1985). The possible role of correcting cataract-induced refractive error to delay the need for surgery should be explored. Subjects • 98 elderly subjects (mean age 67 ± 8 years) were recruited. • 77 subjects had one type of morphological cataract: 34 subjects had cortical cataract, 21 had nuclear cataract and 21 had posterior subcapsular cataract (PSC). • 22 subjects had normal, healthy eyes. • Ocular screening excluded amblyopia, strabismus, eye disease or surgery. -

Treatment of Congenital Ptosis
13 Review Article Page 1 of 13 Treatment of congenital ptosis Vladimir Kratky1,2^ 1Department of Ophthalmology, Queen’s University, Kingston, Canada; 21st Medical Faculty, Charles University, Prague, Czech Republic Correspondence to: Vladimir Kratky, BSc, MD, FRCSC, DABO. Associate Professor of Ophthalmology, Director of Ophthalmic Plastic and Orbital Surgery, Oculoplastics Fellowship Director, Queen’s University, Kingston, Canada; 1st Medical Faculty, Charles University, Prague, Czech Republic. Email: [email protected]. Abstract: Congenital ptosis is an abnormally low position of the upper eyelid, with respect to the visual axis in the primary gaze. It can be present at birth or manifest itself during the first year of life and can be bilateral or unilateral. Additionally, it may be an isolated finding or part of a constellation of signs of a specific syndrome or systemic associations. Depending on how much it interferes with the visual axis, it may be considered as a functional or a cosmetic condition. In childhood, functional ptosis can lead to deprivation amblyopia and astigmatism and needs to be treated. However, even mild ptosis with normal vision can lead to psychosocial problems and correction is also advised, albeit on a less urgent basis. Although, patching and glasses can be prescribed to treat the amblyopia, the mainstay of management is surgical. There are several types of surgical procedure available depending on the severity and etiology of the droopy eyelid. The first part of this paper will review the different categories of congenital ptosis, including more common associated syndromes. The latter part will briefly cover the different surgical approaches, with emphasis on how to choose the correct condition. -

Fact Sheet: Refractive Errors
Fact Sheet: Refractive Errors More than 11 million Americans have common vision problems that can be corrected with the use of prescriptive eyewear such as glasses or contact lenses.1 These conditions are known as refractive errors and they occur when the eye doesn’t correctly bend, or ―refract,‖ light as it enters the eye. Common refractive errors include the following: o Nearsightedness (also called myopia)—A condition where objects up close appear clearly, while objects far away appear blurry. With nearsightedness, light comes to focus in front of the retina instead of on the retina. o Farsightedness (also called hyperopia)—A common type of refractive error where distant objects may be seen more clearly than objects that are near. However, people experience farsightedness differently. Some people may not notice any problems with their vision, especially when they are young. For people with significant farsightedness, vision can be blurry for objects at any distance, near or far. o Astigmatism—A condition in which the eye does not focus light evenly onto the retina, the light-sensitive tissue at the back of the eye. This can cause images to appear blurry and stretched out. o Presbyopia—An age-related condition in which the ability to focus up close becomes more difficult. As the eye ages, the lens can no longer change shape enough to allow the eye to focus close objects clearly. Refractive errors are one of the most common—and correctable—causes of visual impairment in the United States. According to a recent study led by the National Eye Institute (NEI), approximately half of all American adults don’t have the 20/20 vision physicians consider optimal due to refractive errors.2 Women experience refractive error more frequently than men: Twenty-six percent more women aged 12 and older have uncorrected visual impairment due to refractive error compared with men aged 12 and older. -

Recessive Mutations of the Gene TRPM1 Abrogate on Bipolar Cell Function and Cause Complete Congenital Stationary Night Blindness in Humans
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Elsevier - Publisher Connector REPORT Recessive Mutations of the Gene TRPM1 Abrogate ON Bipolar Cell Function and Cause Complete Congenital Stationary Night Blindness in Humans Zheng Li,1 Panagiotis I. Sergouniotis,1 Michel Michaelides,1,2 Donna S. Mackay,1 Genevieve A. Wright,2 Sophie Devery,2 Anthony T. Moore,1,2 Graham E. Holder,1,2 Anthony G. Robson,1,2 and Andrew R. Webster1,2,* Complete congenital stationary night blindness (cCSNB) is associated with loss of function of rod and cone ON bipolar cells in the mammalian retina. In humans, mutations in NYX and GRM6 have been shown to cause the condition. Through the analysis of a consan- guineous family and screening of nine additional pedigrees, we have identified three families with recessive mutations in the gene TRPM1 encoding transient receptor potential cation channel, subfamily M, member 1, also known as melastatin. A number of other variants of unknown significance were found. All patients had myopia, reduced central vision, nystagmus, and electroretinographic evidence of ON bipolar cell dysfunction. None had abnormalities of skin pigmentation, although other skin conditions were reported. RNA derived from human retina and skin was analyzed and alternate 50 exons were determined. The most 50 exon is likely to harbor an initiation codon, and the protein sequence is highly conserved across vertebrate species. These findings suggest an important role of this specific cation channel for the normal function of ON bipolar cells in the human retina. Congenital stationary night blindness (CSNB) is a group of of the gene encoding transient receptor potential cation genetically determined, nondegenerative disorders of the channel, subfamily M, member 1 (TRPM1 [MIM *603576]) retina associated with lifelong deficient vision in the dark has been discovered in the skin and retina of horses homo- and often nystagmus and myopia.