Original Article
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Conserved DNMT1-Dependent Methylation Regions in Human Cells
Freeman et al. Epigenetics & Chromatin (2020) 13:17 https://doi.org/10.1186/s13072-020-00338-8 Epigenetics & Chromatin RESEARCH Open Access The conserved DNMT1-dependent methylation regions in human cells are vulnerable to neurotoxicant rotenone exposure Dana M. Freeman1 , Dan Lou1, Yanqiang Li1, Suzanne N. Martos1 and Zhibin Wang1,2,3* Abstract Background: Allele-specifc DNA methylation (ASM) describes genomic loci that maintain CpG methylation at only one inherited allele rather than having coordinated methylation across both alleles. The most prominent of these regions are germline ASMs (gASMs) that control the expression of imprinted genes in a parent of origin-dependent manner and are associated with disease. However, our recent report reveals numerous ASMs at non-imprinted genes. These non-germline ASMs are dependent on DNA methyltransferase 1 (DNMT1) and strikingly show the feature of random, switchable monoallelic methylation patterns in the mouse genome. The signifcance of these ASMs to human health has not been explored. Due to their shared allelicity with gASMs, herein, we propose that non-tradi- tional ASMs are sensitive to exposures in association with human disease. Results: We frst explore their conservancy in the human genome. Our data show that our putative non-germline ASMs were in conserved regions of the human genome and located adjacent to genes vital for neuronal develop- ment and maturation. We next tested the hypothesized vulnerability of these regions by exposing human embryonic kidney cell HEK293 with the neurotoxicant rotenone for 24 h. Indeed,14 genes adjacent to our identifed regions were diferentially expressed from RNA-sequencing. We analyzed the base-resolution methylation patterns of the predicted non-germline ASMs at two neurological genes, HCN2 and NEFM, with potential to increase the risk of neurodegenera- tion. -

Prediction of Human Disease Genes by Human-Mouse Conserved Coexpression Analysis
Prediction of Human Disease Genes by Human-Mouse Conserved Coexpression Analysis Ugo Ala1., Rosario Michael Piro1., Elena Grassi1, Christian Damasco1, Lorenzo Silengo1, Martin Oti2, Paolo Provero1*, Ferdinando Di Cunto1* 1 Molecular Biotechnology Center, Department of Genetics, Biology and Biochemistry, University of Turin, Turin, Italy, 2 Department of Human Genetics and Centre for Molecular and Biomolecular Informatics, University Medical Centre Nijmegen, Nijmegen, The Netherlands Abstract Background: Even in the post-genomic era, the identification of candidate genes within loci associated with human genetic diseases is a very demanding task, because the critical region may typically contain hundreds of positional candidates. Since genes implicated in similar phenotypes tend to share very similar expression profiles, high throughput gene expression data may represent a very important resource to identify the best candidates for sequencing. However, so far, gene coexpression has not been used very successfully to prioritize positional candidates. Methodology/Principal Findings: We show that it is possible to reliably identify disease-relevant relationships among genes from massive microarray datasets by concentrating only on genes sharing similar expression profiles in both human and mouse. Moreover, we show systematically that the integration of human-mouse conserved coexpression with a phenotype similarity map allows the efficient identification of disease genes in large genomic regions. Finally, using this approach on 850 OMIM loci characterized by an unknown molecular basis, we propose high-probability candidates for 81 genetic diseases. Conclusion: Our results demonstrate that conserved coexpression, even at the human-mouse phylogenetic distance, represents a very strong criterion to predict disease-relevant relationships among human genes. Citation: Ala U, Piro RM, Grassi E, Damasco C, Silengo L, et al. -

The Conserved DNMT1 Dependent Methylation Regions in Human Cells Are Vulnerable to Environmental Rotenone
bioRxiv preprint doi: https://doi.org/10.1101/798587; this version posted October 9, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. The conserved DNMT1 dependent methylation regions in human cells are vulnerable to environmental rotenone. Dana M. Freemana, Dan Loua, Yanqiang Lia, Suzanne N. Martosa, Zhibin Wanga* aLaboratory of Environmental Epigenomes, Department of Environmental Health & Engineering, Bloomberg School of Public Health, Johns Hopkins University, Baltimore, MD *To whom correspondence should be addressed: Zhibin Wang, Ph.D., Associate Professor, Laboratory of Environmental Epigenomes, Department of Environmental Health and Engineering, Bloomberg School of Public Health, Johns Hopkins University, Baltimore, MD. Phone: (410) 955-7840; Email: [email protected] Abstract Allele-specific DNA methylation (ASM) describes genomic loci that maintain CpG methylation at only one inherited allele rather than having coordinated methylation across both alleles. The most prominent of these regions are germline ASMs (gASMs) that control the expression of imprinted genes in a parent of origin- dependent manner and are associated with disease. However, our recent report reveals numerous ASMs at non-imprinted genes. These non-germline ASMs are dependent on DNA methyltransferase 1 (DNMT1) and strikingly show the feature of random, switchable monoallelic methylation patterns in the mouse genome. The significance of these ASMs to human health has not been explored. Due to their shared allelicity with gASMs, herein, we propose that non-traditional ASMs are sensitive to exposures in association with human disease. We first explore their conservancy in the human genome. -

Content Based Search in Gene Expression Databases and a Meta-Analysis of Host Responses to Infection
Content Based Search in Gene Expression Databases and a Meta-analysis of Host Responses to Infection A Thesis Submitted to the Faculty of Drexel University by Francis X. Bell in partial fulfillment of the requirements for the degree of Doctor of Philosophy November 2015 c Copyright 2015 Francis X. Bell. All Rights Reserved. ii Acknowledgments I would like to acknowledge and thank my advisor, Dr. Ahmet Sacan. Without his advice, support, and patience I would not have been able to accomplish all that I have. I would also like to thank my committee members and the Biomed Faculty that have guided me. I would like to give a special thanks for the members of the bioinformatics lab, in particular the members of the Sacan lab: Rehman Qureshi, Daisy Heng Yang, April Chunyu Zhao, and Yiqian Zhou. Thank you for creating a pleasant and friendly environment in the lab. I give the members of my family my sincerest gratitude for all that they have done for me. I cannot begin to repay my parents for their sacrifices. I am eternally grateful for everything they have done. The support of my sisters and their encouragement gave me the strength to persevere to the end. iii Table of Contents LIST OF TABLES.......................................................................... vii LIST OF FIGURES ........................................................................ xiv ABSTRACT ................................................................................ xvii 1. A BRIEF INTRODUCTION TO GENE EXPRESSION............................. 1 1.1 Central Dogma of Molecular Biology........................................... 1 1.1.1 Basic Transfers .......................................................... 1 1.1.2 Uncommon Transfers ................................................... 3 1.2 Gene Expression ................................................................. 4 1.2.1 Estimating Gene Expression ............................................ 4 1.2.2 DNA Microarrays ...................................................... -

Peripheral Nerve Single-Cell Analysis Identifies Mesenchymal Ligands That Promote Axonal Growth
Research Article: New Research Development Peripheral Nerve Single-Cell Analysis Identifies Mesenchymal Ligands that Promote Axonal Growth Jeremy S. Toma,1 Konstantina Karamboulas,1,ª Matthew J. Carr,1,2,ª Adelaida Kolaj,1,3 Scott A. Yuzwa,1 Neemat Mahmud,1,3 Mekayla A. Storer,1 David R. Kaplan,1,2,4 and Freda D. Miller1,2,3,4 https://doi.org/10.1523/ENEURO.0066-20.2020 1Program in Neurosciences and Mental Health, Hospital for Sick Children, 555 University Avenue, Toronto, Ontario M5G 1X8, Canada, 2Institute of Medical Sciences University of Toronto, Toronto, Ontario M5G 1A8, Canada, 3Department of Physiology, University of Toronto, Toronto, Ontario M5G 1A8, Canada, and 4Department of Molecular Genetics, University of Toronto, Toronto, Ontario M5G 1A8, Canada Abstract Peripheral nerves provide a supportive growth environment for developing and regenerating axons and are es- sential for maintenance and repair of many non-neural tissues. This capacity has largely been ascribed to paracrine factors secreted by nerve-resident Schwann cells. Here, we used single-cell transcriptional profiling to identify ligands made by different injured rodent nerve cell types and have combined this with cell-surface mass spectrometry to computationally model potential paracrine interactions with peripheral neurons. These analyses show that peripheral nerves make many ligands predicted to act on peripheral and CNS neurons, in- cluding known and previously uncharacterized ligands. While Schwann cells are an important ligand source within injured nerves, more than half of the predicted ligands are made by nerve-resident mesenchymal cells, including the endoneurial cells most closely associated with peripheral axons. At least three of these mesen- chymal ligands, ANGPT1, CCL11, and VEGFC, promote growth when locally applied on sympathetic axons. -

CRAVAT and Mupit Interacsve: Web Tools for Cancer Mutason Analysis
CRAVAT and MuPIT Interac2ve: Web Tools for Cancer Mutaon Analysis Rachel Karchin, Ph.D. Department of Biomedical Engineering Ins2tute for Computaonal Medicine Johns Hopkins University The Cancer Genome Atlas’ 2nd Annual Scien2fic Symposium November 27-28, 2012 Need for computaonal tools to analyze large-scale cancer mutaon data 2 Goal is to provide an end-to-end mutaon analysis workflow List of mutaons from tumor sequencing Iden2fy type of Iden2fy known Map to transcripts change (missense, variants and nonsense, silent) mutaons Analysis Predict driver vs. Predict func2onal Visualize Find significantly random impact of mutaons on mutated genes mutaons mutaons ter2ary structure and pathways 3 The majority of somac mutaons in tumor exomes are missense Sjoblom et al 2006 Hunter et al 2005 Davies et al 2005 Stephens et al 2005 Sjoblom et al 2006 Missense Nonsense Silent Splice Other Colorectal Breast Greenman et al 2007 Jones et al 2008 Parsons et al 2008 McLendon et al 2008 Ding et al 2008 Parsons et al 2011 Jones et al 2010 TCGA 2010 Stransky et al 2011 Li et al 2011 4 hfp://www.cravat.us Tools for evaluang missense mutaons – CHASM: cancer driver analysis hfp://mupit.icm.jhu.edu – VEST: Func2onal effect analysis – Annotaons (1000g, ESP6500, COSMIC, GeneCards, PubMed) Interac2ve visualizaon on 3D protein structure – Automac mapping onto available structures – Simple interac2ve interface – UniProtKB feature table annotaons provided – Publicaon quality figures 5 Outline • Introduc2on to CRAVAT • Introduc2on to MuPIT • Future plans • Your input 6 -

The LGI1 Gene Involved in Lateral Temporal Lobe Epilepsy Belongs to a New Subfamily of Leucine-Rich Repeat Proteins
FEBS 26082 FEBS Letters 519 (2002) 71^76 The LGI1 gene involved in lateral temporal lobe epilepsy belongs to a new subfamily of leucine-rich repeat proteins Wenli Gua, Andrea Weversb, Hannsjo«rg Schro«derb, Karl-Heinz Grzeschikc, Christian Derstd, Eylert Brodtkorbe, Rob de Vosf , Ortrud K. Steinleina;Ã aInstitute of Human Genetics, University Hospital Bonn, Wilhelmstrasse 31, D-53111 Bonn, Germany bInstitute for Anatomy II, Neuroanatomy, University of Cologne, Joseph-Stelzmann Strasse 9, 50931 Cologne, Germany cDepartment of Human Genetics, University of Marburg, Bahnhofstrasse 7, 35037 Marburg, Germany dInstitute of Physiology II, University of Freiburg, Herman Herder Strasse 7, 79104 Freiburg, Germany eDepartment of Neurology, Trondheim University Hospital, N-7006 Trondheim, Norway f Laboratorium Pathologie Oost Nederland, Burgemeester Edo Bergsmalaan 1, 7512 AD Enschede, The Netherlands Received 14 March 2002; revised 10 April 2002; accepted 12 April 2002 First published online 25 April 2002 Edited by Guido Tettamanti cell line, and was therefore named according to its possible Abstract Recently mutations in the LGI1 (leucine-rich, glioma- inactivated 1) gene have been found in human temporal lobe function as a tumor-suppressor gene. The possible role in epilepsy. We have now identified three formerly unknown LGI- tumorigenesis was further supported by the observation of like genes. Hydropathy plots and pattern analysis showed that LGI1 down-regulation in malignant gliomas [2]. Thus it LGI genes encode proteins with large extra- and intracellular came as a surprise that LGI1 is associated with epilepsy in domains connected by a single transmembrane region. Sequence several families not known for an unusual accumulation of analysis demonstrated that LGI1, LGI2, LGI3, and LGI4 brain tumors or other malignancies [1]. -

Systematic Detection of Brain Protein-Coding Genes Under Positive Selection During Primate Evolution and Their Roles in Cognition
Downloaded from genome.cshlp.org on October 7, 2021 - Published by Cold Spring Harbor Laboratory Press Title: Systematic detection of brain protein-coding genes under positive selection during primate evolution and their roles in cognition Short title: Evolution of brain protein-coding genes in humans Guillaume Dumasa,b, Simon Malesysa, and Thomas Bourgerona a Human Genetics and Cognitive Functions, Institut Pasteur, UMR3571 CNRS, Université de Paris, Paris, (75015) France b Department of Psychiatry, Université de Montreal, CHU Ste Justine Hospital, Montreal, QC, Canada. Corresponding author: Guillaume Dumas Human Genetics and Cognitive Functions Institut Pasteur 75015 Paris, France Phone: +33 6 28 25 56 65 [email protected] Dumas, Malesys, and Bourgeron 1 of 40 Downloaded from genome.cshlp.org on October 7, 2021 - Published by Cold Spring Harbor Laboratory Press Abstract The human brain differs from that of other primates, but the genetic basis of these differences remains unclear. We investigated the evolutionary pressures acting on almost all human protein-coding genes (N=11,667; 1:1 orthologs in primates) based on their divergence from those of early hominins, such as Neanderthals, and non-human primates. We confirm that genes encoding brain-related proteins are among the most strongly conserved protein-coding genes in the human genome. Combining our evolutionary pressure metrics for the protein- coding genome with recent datasets, we found that this conservation applied to genes functionally associated with the synapse and expressed in brain structures such as the prefrontal cortex and the cerebellum. Conversely, several genes presenting signatures commonly associated with positive selection appear as causing brain diseases or conditions, such as micro/macrocephaly, Joubert syndrome, dyslexia, and autism. -
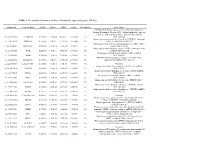
Table 1 the Statistical Metrics for Key Differentially Expressed Genes (Degs)
Table 1 The statistical metrics for key differentially expressed genes (DEGs) Agiliant Id Gene Symbol logFC pValue FDR tvalue Regulation Gene Name PREDICTED: Homo sapiens similar to Filamin-C (Gamma- filamin) (Filamin-2) (Protein FLNc) (Actin-binding-like protein) (ABP-L) (ABP-280-like protein) (LOC649056), mRNA A_24_P237896 LOC649056 0.509843 9.18E-14 4.54E-11 12.07302 Up [XR_018580] Homo sapiens programmed cell death 10 (PDCD10), transcript A_23_P18325 PDCD10 0.243111 2.8E-12 9.24E-10 10.62808 Up variant 1, mRNA [NM_007217] Homo sapiens Rho GTPase activating protein 27 (ARHGAP27), A_23_P141335 ARHGAP27 0.492709 3.97E-12 1.22E-09 10.48571 Up mRNA [NM_199282] Homo sapiens tubby homolog (mouse) (TUB), transcript variant A_23_P53110 TUB 0.528219 1.77E-11 4.56E-09 9.891033 Up 1, mRNA [NM_003320] Homo sapiens MyoD family inhibitor (MDFI), mRNA A_23_P42168 MDFI 0.314474 1.81E-10 3.74E-08 8.998697 Up [NM_005586] PREDICTED: Homo sapiens hypothetical LOC644701 A_32_P56890 LOC644701 0.444703 3.6E-10 7.09E-08 8.743973 Up (LOC644701), mRNA [XM_932316] A_32_P167111 A_32_P167111 0.873588 7.41E-10 1.4E-07 8.47781 Up Unknown Homo sapiens zinc finger protein 784 (ZNF784), mRNA A_24_P221424 ZNF784 0.686781 9.18E-10 1.68E-07 8.399687 Up [NM_203374] Homo sapiens lin-28 homolog (C. elegans) (LIN28), mRNA A_23_P74895 LIN28 0.218876 1.27E-09 2.24E-07 8.282224 Up [NM_024674] Homo sapiens ribosomal protein L5 (RPL5), mRNA A_23_P12140 RPL5 0.247598 1.81E-09 3.11E-07 8.154317 Up [NM_000969] Homo sapiens cDNA FLJ43841 fis, clone TESTI4006137. -

World Journal of Cardiology
World Journal of W J C Cardiology Submit a Manuscript: http://www.f6publishing.com World J Cardiol 2017 April 26; 9(4): 320-331 DOI: 10.4330/wjc.v9.i4.320 ISSN 1949-8462 (online) ORIGINAL ARTICLE Basic Study Dissection of Z-disc myopalladin gene network involved in the development of restrictive cardiomyopathy using system genetics approach Qingqing Gu, Uzmee Mendsaikhan, Zaza Khuchua, Byron C Jones, Lu Lu, Jeffrey A Towbin, Biao Xu, Enkhsaikhan Purevjav Qingqing Gu, Biao Xu, Department of Cardiology, Drum Tower (LL), R01 HL53392 and R01 HL087000 (JAT). Clinic Hospital, Nanjing Medical University, Nanjing 211166, Jiangsu Province, China Institutional animal care and use committee statement: All animal studies were approved by institutional IACUC of the Qingqing Gu, Lu Lu, Department of Genetics, Genomics and University of Tennessee Health Science Center (UTHSC). Informatics, University of Tennessee Health Science Center, Memphis, TN 38103, United States Conflict-of-interest statement: To the best of our knowledge, no conflict of interest exists. Uzmee Mendsaikhan, the Heart Institute, Cincinnati Child- ren’s Hospital Medical Center and Mongolian National University Data sharing statement: Resources: Principles and Guidelines of Medical Sciences, Ulaanbaatar 14210, Mongolia for Recipients of NIH Grants and Contracts” issued in December, 1999. Dr. Lu Lu is responsible for coordinating data sharing Zaza Khuchua, the Heart Institute, Cincinnati Children’s Hospital through GeneNetwork (GN) at the: http://www.genenetwork. Medical Center, Cincinnati, OH 45229, United States org/webqtl/main.py. GN is a group of linked data sets and tools used to study complex networks of genes, molecules, and higher Byron C Jones, the Neuroscience Institute, Department of order gene function and phenotypes. -

Interactions of Bartonella Henselae with Myeloid Angiogenic Cells and Consequences for Pathological Angiogenesis
Interactions of Bartonella henselae with Myeloid Angiogenic Cells and Consequences for Pathological Angiogenesis Dissertation zur Erlangung des Doktorgrades der Naturwissenschaften vorgelegt beim Fachbereich Biowissenschaften der Johann Wolfgang Goethe-Universität in Frankfurt am Main von Fiona OʼRourke aus Calgary (Kanada) Frankfurt am Main 2015 vom Fachbereich Biowissenschaften der Johann Wolfgang Goethe-Universität als Dissertation angenommen. Dekanin: Prof. Dr. Meike Piepenbring 1. Gutachter: Prof. Dr. Volker Müller 2. Gutachter: Prof. Dr. Volkhard A. J. Kempf Datum der Disputation: 02.12.2015 I Table of Contents 1. Introduction .................................................................................................... 1 1.1 Bartonella ................................................................................................................................1 1.2 Bartonella henselae ..................................................................................................................2 1.3 Infection-associated pathological angiogenesis ..........................................................................2 1.4 B. henselae pathogenicity strategy ............................................................................................4 1.4.1 Mitogenic stimulus ....................................................................................................................... 6 1.4.2 Inhibtion of apoptosis ................................................................................................................. -

Copy Number Networks to Guide Combinatorial Therapy for Cancer and Other Disorders
bioRxiv preprint doi: https://doi.org/10.1101/005942; this version posted June 24, 2014. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. Copy number networks to guide combinatorial therapy for cancer and other disorders Andy Lin and Desmond J. Smith1 Department of Molecular and Medical Pharmacology David Geffen School of Medicine UCLA 23-120 CHS, Box 951735 Los Angeles, CA 90095-1735 USA 1Corresponding author Tel: 310-206-0086 Fax: 310-825-6267 [email protected] bioRxiv preprint doi: https://doi.org/10.1101/005942; this version posted June 24, 2014. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. CNA networks and combinatorial therapy ABSTRACT The dwindling drug pipeline is driving increased interest in the use of genome datasets to inform drug treatment. In particular, networks based on transcript data and protein-protein interactions have been used to design therapies that employ drug combinations. But there has been less focus on employing human genetic interaction networks constructed from copy number alterations (CNAs). These networks can be charted with sensitivity and precision by seeking gene pairs that tend to be amplified and/or deleted in tandem, even when they are located at a distance on the genome.