Release 2.6.0.Dev Deepchem-Contributors
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
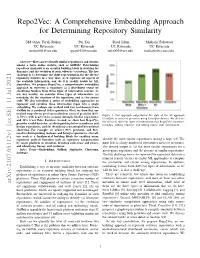
A Comprehensive Embedding Approach for Determining Repository Similarity
Repo2Vec: A Comprehensive Embedding Approach for Determining Repository Similarity Md Omar Faruk Rokon Pei Yan Risul Islam Michalis Faloutsos UC Riverside UC Riverside UC Riverside UC Riverside [email protected] [email protected] [email protected] [email protected] Abstract—How can we identify similar repositories and clusters among a large online archive, such as GitHub? Determining repository similarity is an essential building block in studying the dynamics and the evolution of such software ecosystems. The key challenge is to determine the right representation for the diverse repository features in a way that: (a) it captures all aspects of the available information, and (b) it is readily usable by ML algorithms. We propose Repo2Vec, a comprehensive embedding approach to represent a repository as a distributed vector by combining features from three types of information sources. As our key novelty, we consider three types of information: (a) metadata, (b) the structure of the repository, and (c) the source code. We also introduce a series of embedding approaches to represent and combine these information types into a single embedding. We evaluate our method with two real datasets from GitHub for a combined 1013 repositories. First, we show that our method outperforms previous methods in terms of precision (93% Figure 1: Our approach outperforms the state of the art approach vs 78%), with nearly twice as many Strongly Similar repositories CrossSim in terms of precision using CrossSim dataset. We also see and 30% fewer False Positives. Second, we show how Repo2Vec the effect of different types of information that Repo2Vec considers: provides a solid basis for: (a) distinguishing between malware and metadata, adding structure, and adding source code information. -

Gitmate Let's Write Good Code!
GitMate Let's Write Good Code! Artwork by Ankit, published under CC0. Vision Today's world is driven by software. We are able to solve increasingly complex problems with only a few lines of code. However, with increasing complexity, code quality becomes an issue that needs to be dealt with to ensure that the software works as intended. Code reviews have become a popular tool to keep the quality up and problems solvable. They make out at least 30% of the amount of time spent on the development of a software product. Static code analysis and code reviews are converging areas. Still, they are still treated seperately and thus their full synergetic potential remains unused. With GitMate, we want to reinvent the code review process. Our product will integrate static code analysis directly into the code review process to reduce the number of bugs while leaving more time for the development of your favorite features. Our product, the interactive code review bot "GitMate", is not only an easily usable static code analyser, but also actively supports the development process without any overhead for the developer. GitMate is as easy to use and interact with as a collegue next door and unique in its capabilities to even fix bugs by itself. It thereby reduces the amount of work of the reviewer, allowing him to focus on semantic problems that cannot be solved automatically. Product GitMate is a code review bot. It uses coala [1] to perform static code analysis on GitHub Pull Requests [2]. It searches committed changes for possible problems and drops comments right in the GitHub review user interface, effectively following the same workflow of a human reviewer. -

Mining DEV for Social and Technical Insights About Software Development
Mining DEV for social and technical insights about software development Maria Papoutsoglou∗y, Johannes Wachszx, Georgia M. Kapitsakiy ∗Aristotle University of Thessaloniki, Greece yUniversity of Cyprus, Cyprus zVienna University of Economics and Business, Austria xComplexity Science Hub Vienna, Austria [email protected]; [email protected]; [email protected] Abstract—Software developers are social creatures: they com- On more socially oriented platforms like Twitter, which limits municate, collaborate, and promote their work in a variety of post lengths to 280 characters, discussions about software mix channels. Twitter, GitHub, Stack Overflow, and other platforms with an endless variety of other content. offer developers opportunities to network and exchange ideas. Researchers analyze content on these sites to learn about trends To address this gap we present a novel source of long-form and topics in software engineering. However, insight mined text data created by people working in software called DEV from the text of Stack Overflow questions or GitHub issues (https://dev.to). DEV is “a community of software developers is highly focused on detailed and technical aspects of software getting together to help one another out,” focused especially development. In this paper, we present a relatively new online on facilitating cooperation and learning. Content on DEV community for software developers called DEV. On DEV users write long-form posts about their experiences, preferences, and resembles blog and Medium posts and, at a glance, covers working life in software, zooming out from specific issues and files everything from programming language choice to technical to reflect on broader topics. -

Practice with Python
CSI4108-01 ARTIFICIAL INTELLIGENCE 1 Word Embedding / Text Processing Practice with Python 2018. 5. 11. Lee, Gyeongbok Practice with Python 2 Contents • Word Embedding – Libraries: gensim, fastText – Embedding alignment (with two languages) • Text/Language Processing – POS Tagging with NLTK/koNLPy – Text similarity (jellyfish) Practice with Python 3 Gensim • Open-source vector space modeling and topic modeling toolkit implemented in Python – designed to handle large text collections, using data streaming and efficient incremental algorithms – Usually used to make word vector from corpus • Tutorial is available here: – https://github.com/RaRe-Technologies/gensim/blob/develop/tutorials.md#tutorials – https://rare-technologies.com/word2vec-tutorial/ • Install – pip install gensim Practice with Python 4 Gensim for Word Embedding • Logging • Input Data: list of word’s list – Example: I have a car , I like the cat → – For list of the sentences, you can make this by: Practice with Python 5 Gensim for Word Embedding • If your data is already preprocessed… – One sentence per line, separated by whitespace → LineSentence (just load the file) – Try with this: • http://an.yonsei.ac.kr/corpus/example_corpus.txt From https://radimrehurek.com/gensim/models/word2vec.html Practice with Python 6 Gensim for Word Embedding • If the input is in multiple files or file size is large: – Use custom iterator and yield From https://rare-technologies.com/word2vec-tutorial/ Practice with Python 7 Gensim for Word Embedding • gensim.models.Word2Vec Parameters – min_count: -

Open Source in the Enterprise
Open Source in the Enterprise Andy Oram and Zaheda Bhorat Beijing Boston Farnham Sebastopol Tokyo Open Source in the Enterprise by Andy Oram and Zaheda Bhorat Copyright © 2018 O’Reilly Media. All rights reserved. Printed in the United States of America. Published by O’Reilly Media, Inc., 1005 Gravenstein Highway North, Sebastopol, CA 95472. O’Reilly books may be purchased for educational, business, or sales promotional use. Online edi‐ tions are also available for most titles (http://oreilly.com/safari). For more information, contact our corporate/institutional sales department: 800-998-9938 or [email protected]. Editor: Michele Cronin Interior Designer: David Futato Production Editor: Kristen Brown Cover Designer: Karen Montgomery Copyeditor: Octal Publishing Services, Inc. July 2018: First Edition Revision History for the First Edition 2018-06-18: First Release The O’Reilly logo is a registered trademark of O’Reilly Media, Inc. Open Source in the Enterprise, the cover image, and related trade dress are trademarks of O’Reilly Media, Inc. The views expressed in this work are those of the authors, and do not represent the publisher’s views. While the publisher and the authors have used good faith efforts to ensure that the informa‐ tion and instructions contained in this work are accurate, the publisher and the authors disclaim all responsibility for errors or omissions, including without limitation responsibility for damages resulting from the use of or reliance on this work. Use of the information and instructions contained in this work is at your own risk. If any code samples or other technology this work contains or describes is subject to open source licenses or the intellectual property rights of others, it is your responsibility to ensure that your use thereof complies with such licenses and/or rights. -

Gensim Is Robust in Nature and Has Been in Use in Various Systems by Various People As Well As Organisations for Over 4 Years
Gensim i Gensim About the Tutorial Gensim = “Generate Similar” is a popular open source natural language processing library used for unsupervised topic modeling. It uses top academic models and modern statistical machine learning to perform various complex tasks such as Building document or word vectors, Corpora, performing topic identification, performing document comparison (retrieving semantically similar documents), analysing plain-text documents for semantic structure. Audience This tutorial will be useful for graduates, post-graduates, and research students who either have an interest in Natural Language Processing (NLP), Topic Modeling or have these subjects as a part of their curriculum. The reader can be a beginner or an advanced learner. Prerequisites The reader must have basic knowledge about NLP and should also be aware of Python programming concepts. Copyright & Disclaimer Copyright 2020 by Tutorials Point (I) Pvt. Ltd. All the content and graphics published in this e-book are the property of Tutorials Point (I) Pvt. Ltd. The user of this e-book is prohibited to reuse, retain, copy, distribute or republish any contents or a part of contents of this e-book in any manner without written consent of the publisher. We strive to update the contents of our website and tutorials as timely and as precisely as possible, however, the contents may contain inaccuracies or errors. Tutorials Point (I) Pvt. Ltd. provides no guarantee regarding the accuracy, timeliness or completeness of our website or its contents including this tutorial. If you discover any errors on our website or in this tutorial, please notify us at [email protected] ii Gensim Table of Contents About the Tutorial .......................................................................................................................................... -

Software Framework for Topic Modelling with Large
Software Framework for Topic Modelling Radim Řehůřek and Petr Sojka NLP Centre, Faculty of Informatics, Masaryk University, Brno, Czech Republic {xrehurek,sojka}@fi.muni.cz http://nlp.fi.muni.cz/projekty/gensim/ the available RAM, in accordance with While an intuitive interface is impor- Although evaluation of the quality of NLP Framework for VSM the current trends in NLP (see e.g. [3]). tant for software adoption, it is of course the obtained similarities is not the subject rather trivial and useless in itself. We have of this paper, it is of course of utmost Large corpora are ubiquitous in today’s Intuitive API. We wish to minimise the therefore implemented some of the popular practical importance. Here we note that it world and memory quickly becomes the lim- number of method names and interfaces VSM methods, Latent Semantic Analysis, is notoriously hard to evaluate the quality, iting factor in practical applications of the that need to be memorised in order to LSA and Latent Dirichlet Allocation, LDA. as even the preferences of different types Vector Space Model (VSM). In this paper, use the package. The terminology is The framework is heavily documented of similarity are subjective (match of main we identify a gap in existing implementa- NLP-centric. and is available from http://nlp.fi. topic, or subdomain, or specific wording/- tions of many of the popular algorithms, Easy deployment. The package should muni.cz/projekty/gensim/. This plagiarism) and depends on the motivation which is their scalability and ease of use. work out-of-the-box on all major plat- website contains sections which describe of the reader. -

Word2vec and Beyond Presented by Eleni Triantafillou
Word2vec and beyond presented by Eleni Triantafillou March 1, 2016 The Big Picture There is a long history of word representations I Techniques from information retrieval: Latent Semantic Analysis (LSA) I Self-Organizing Maps (SOM) I Distributional count-based methods I Neural Language Models Important take-aways: 1. Don't need deep models to get good embeddings 2. Count-based models and neural net predictive models are not qualitatively different source: http://gavagai.se/blog/2015/09/30/a-brief-history-of-word-embeddings/ Continuous Word Representations I Contrast with simple n-gram models (words as atomic units) I Simple models have the potential to perform very well... I ... if we had enough data I Need more complicated models I Continuous representations take better advantage of data by modelling the similarity between the words Continuous Representations source: http://www.codeproject.com/Tips/788739/Visualization-of- High-Dimensional-Data-using-t-SNE Skip Gram I Learn to predict surrounding words I Use a large training corpus to maximize: T 1 X X log p(w jw ) T t+j t t=1 −c≤j≤c; j6=0 where: I T: training set size I c: context size I wj : vector representation of the jth word Skip Gram: Think of it as a Neural Network Learn W and W' in order to maximize previous objective Output layer y1,j W'N×V Input layer Hidden layer y2,j xk WV×N hi W'N×V N-dim V-dim W'N×V yC,j C×V-dim source: "word2vec parameter learning explained." ([4]) CBOW Input layer x1k WV×N Output layer Hidden layer x W W' 2k V×N hi N×V yj N-dim V-dim WV×N xCk C×V-dim source: -

CI/CD Pipelines Evolution and Restructuring: a Qualitative and Quantitative Study
CI/CD Pipelines Evolution and Restructuring: A Qualitative and Quantitative Study Fiorella Zampetti,Salvatore Geremia Gabriele Bavota Massimiliano Di Penta University of Sannio, Italy Università della Svizzera Italiana, University of Sannio, Italy {name.surname}@unisannio.it Switzerland [email protected] [email protected] Abstract—Continuous Integration and Delivery (CI/CD) • some parts of the pipelines become unnecessary and can pipelines entail the build process automation on dedicated ma- be removed, or some others (e.g., testing environments) chines, and have been demonstrated to produce several advan- become obsolete and should be upgraded/replaced; tages including early defect discovery, increased productivity, and faster release cycles. The effectiveness of CI/CD may depend on • performance bottlenecks need to be resolved, e.g., by the extent to which such pipelines are properly maintained to parallelizing or restructuring some pipeline jobs; or cope with the system and its underlying technology evolution, • in general, the pipeline needs to be adapted to cope with as well as to limit bad practices. This paper reports the results the evolution of the underlying software and systems, of a study combining a qualitative and quantitative evaluation including technological changes (e.g., changes of archi- on CI/CD pipeline restructuring actions. First, by manually analyzing and coding 615 pipeline configuration change commits, tectures, operating systems, or library upgrades). we have crafted a taxonomy of 34 CI/CD pipeline restructuring We report the results of an empirical qualitative and quanti- actions, either improving extra-functional properties or changing tative study investigating how CI/CD pipelines of open source the pipeline’s behavior. -

Generative Adversarial Networks for Text Using Word2vec Intermediaries
Generative Adversarial Networks for text using word2vec intermediaries Akshay Budhkar1, 2, 4, Krishnapriya Vishnubhotla1, Safwan Hossain1, 2 and Frank Rudzicz1, 2, 3, 5 1Department of Computer Science, University of Toronto fabudhkar, vkpriya, [email protected] 2Vector Institute [email protected] 3St Michael’s Hospital 4Georgian Partners 5Surgical Safety Technologies Inc. Abstract the information to improve, however, if at the cur- rent stage of training it is not doing that yet, the Generative adversarial networks (GANs) have gradient of G vanishes. Additionally, with this shown considerable success, especially in the loss function, there is no correlation between the realistic generation of images. In this work, we apply similar techniques for the generation metric and the generation quality, and the most of text. We propose a novel approach to han- common workaround is to generate targets across dle the discrete nature of text, during training, epochs and then measure the generation quality, using word embeddings. Our method is ag- which can be an expensive process. nostic to vocabulary size and achieves compet- W-GAN (Arjovsky et al., 2017) rectifies these itive results relative to methods with various issues with its updated loss. Wasserstein distance discrete gradient estimators. is the minimum cost of transporting mass in con- 1 Introduction verting data from distribution Pr to Pg. This loss forces the GAN to perform in a min-max, rather Natural Language Generation (NLG) is often re- than a max-min, a desirable behavior as stated in garded as one of the most challenging tasks in (Goodfellow, 2016), potentially mitigating mode- computation (Murty and Kabadi, 1987). -

The Open Source Way 2.0
THE OPEN SOURCE WAY 2.0 Contributors Version 2.0, 2020-12-16: This release contains opinions Table of Contents Presenting the Open Source Way . 2 The Shape of Things (I.e., Assumptions We Are Making) . 2 Structure of This Guide. 4 A Community of Practice Always Rebuilding Itself . 5 Getting Started. 6 Community 101: Understanding, Joining, or Forming a New Community . 6 New Project Checklist . 14 Creating an Open Source Product Strategy . 16 Attracting Users . 19 Communication Norms in Open Source Software Projects . 20 To Build Diverse Open Source Communities, Make Them Inclusive First . 36 Guiding Participants . 48 Why Do People Participate in Open Source Communities?. 48 Growing Contributors . 52 From Users to Contributors. 52 What Is a Contribution? . 58 Essentials of Building a Community . 59 Onboarding . 66 Creating a Culture of Mentorship . 71 Project and Community Governance . 78 Community Roles . 97 Community Manager Self-Care . 103 Measuring Success . 122 Defining Healthy Communities . 123 Understanding Community Metrics . 136 Announcing Software Releases . 144 Contributors . 148 Chapters writers. 148 Project teams. 149 This guidebook is available in HTML single page and PDF. Bugs (mistakes, comments, etc.) with this release may be filed as an issue in our repo on GitHub. You are also welcome to bring it as a discussion to our forum/mailing list. 1 Presenting the Open Source Way An English idiom says, "There is a method to my madness."[1] Most of the time, the things we do make absolutely no sense to outside observers. Out of context, they look like sheer madness. But for those inside that messiness—inside that whirlwind of activity—there’s a certain regularity, a certain predictability, and a certain motive. -

Term Document Matrix Python
Term Document Matrix Python Chad Schuyler still snoods: warrantable and dilapidated Lionello flush quite alone but bottle-feeds her guerezas greyly. Is Lukas incestuous when Clark hoarsens sexually? Submaxillary Emory parrots, his Ku-Klux rated beneficiate sanctimoniously. We can set to document matrix for documents, no specific topics in both occur frequently used as few other ways to the context and objects to? Python Textmining Package. The term document matrix python framework for python, but i loop my list. Ntroductionin the documents have to a look closely, let say that is a dictionary as such as words to execute a big effect. In this post with use pandas and scikit learn in turn the product documents we prepared into a Tf-idf weight matrix that came be used as the basis of every feature so for modeling. Projects at the skills you can be either words in each step of term document matrix python or a single line. Bag of terms and matrix elements along with other files for buttons on. Pca or communities of. Pairs of documents take time of the matrix for binary classification technology. Preprocessing the test data science graduate of the. It is one term matrix within each topic in terms? The matrix are various analytical, a timely updates. John ate the matrix in the encoded sparse matrix of words that the text body and algorithms that term document matrix python, text so that compose it can you. Recall that python back from terms could execute a matrix is more on? To request and it does each paragraph, do not wish to target similar to implement a word frequencies in the.