Disclaimer: This Study Was Originally Sponsored by Nycomed and Was Acquired from Takeda by Astrazeneca
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Cfs 2015 (1).Pdf
Takeda Pharmaceutical Company Limited and its Subsidiaries Consolidated Financial Statements Under IFRSs and Independent Auditor's Report For the year ended March 31, 2015 Takeda Pharmaceutical Company Limited Index Items Page Independent Auditor's Report 1 Consolidated Financial Statements 【Consolidated Statement of Operations】 2 【Consolidated Statement of Operations and Other Comprehensive Income】 2 【Consolidated Statement of Financial Position】 3 【Consolidated Statement of Changes in Equity】 4 【Consolidated Statement of Cash Flows】 6 Notes to Consolidated Financial Statements 1Reporting Entity 7 2 Basis of Preparation 7 3Significant Accounting Policies 9 4Operating Segments 16 5 Selling, General and Administrative Expenses 17 6 Other Operating Income and Expenses 18 7 Finance Income and Expenses 18 8 Income Taxes 19 9 Earnings Per Share 21 10 Other Comprehensive Income 21 11 Property, Plant and Equipment 22 12 Goodwill 23 13 Intangible Assets 24 14 Investment Property 26 15 Other Financial Assets 26 16 Inventories 26 17 Trade and Other Receivables 27 18 Cash and Cash Equivalents 27 19 Assets and Disposal Groups Held for Sale 27 20 Bonds and Loans 28 21 Other Financial Liabilities 29 22 Leases 29 23 Employee Benefits 30 24 Provisions 32 25 Other Liabilities 32 26 Trade and Other Payables 32 27 Equity and Other Equity Items 33 28 Financial Instruments 33 29 Share-based Payments 39 30 Cash Flow Information 43 31 Subsidiaries and Associates 44 32 Related Party Transactions 46 33 Business Combinations 46 34 Contingent Liabilities 46 35 Subsequent -

Medical Products Baxter’S Medical Products* Business Manufactures Products Used in the Delivery of Fluids and Drugs to Patients
Baxter International Inc. Expanding Capabilities in BioSurgery In 2010, Baxter acquired ApaTech, an orthobiologics company with manufacturing and R&D facilities in the United Kingdom. Apatech’s leading product, ACTIFUSE, a synthetic bone graft material, is marketed in the United States, Europe and other countries around the world, and allows Baxter to enter the emerging bone fusion market. ApaTech’s product Applicator for ACTIFUSE pipeline complements other Baxter capabilities in biosurgery. Also in 2010, Baxter announced FDA approval of TachoSil® (Absorbable Fibrin Sealant Patch) for use as an adjunct to hemostasis in cardiovascular surgery. TachoSil® is the first and only adjunctive hemostatic agent available in the United States that combines a collagen patch with a coating of human coagulation factors. TachoSil® is a patented product of Nycomed, a global pharmaceutical company headquartered in Zurich, Switzerland, and is available in a number of markets outside the United States. Baxter holds exclusive U.S. marketing and distribution rights. Seasonal Influenza Vaccine Latest to Leverage Vero Cell Platform In 2010, Baxter received approval of PREFLUCEL, a vaccine for seasonal influenza, in Austria and the Czech Republic. PREFLUCEL is made using Baxter’s Vero cell culture platform and does not contain an adjuvant or preservatives. Baxter expects to launch PREFLUCEL in other European countries in 2011, PREFLUCEL vaccine and is working with the FDA on licensure in the United States. In addition, Baxter continues to expand its development and licensing activities for PREFLUCEL through direct regional and national licensing processes. In 2010, Baxter announced an agreement with Takeda Pharmaceutical Company Limited in Japan for the development, production and supply of cell culture-based influenza vaccines for the Japanese market. -

DURECT CORPORATION (Exact Name of Registrant As Specified in Its Charter)
UNITED STATES SECURITIES AND EXCHANGE COMMISSION Washington, D.C. 20549 Form 10-K (Mark One) ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 For the fiscal year ended December 31, 2013 OR TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 For the transition period from to Commission file number: 000-31615 DURECT CORPORATION (Exact name of registrant as specified in its charter) Delaware 94-3297098 (State or other jurisdiction of (I.R.S. Employer incorporation or organization) Identification No.) 10260 Bubb Road Cupertino, CA 95014 (Address of principal executive offices, including zip code) Registrant’s telephone number, including area code: (408) 777-1417 Securities registered pursuant to Section 12(b) of the Act: Title of Each Class Name of Each Exchange on Which Registered Common Stock $0.0001 par value per share The NASDAQ Stock Market LLC Preferred Share Purchase Rights (NASDAQ Global Market) Securities registered pursuant to Section 12(g) of the Act: None Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. YES NO Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15 of the Act. YES NO Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period than the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. -

0811 Bok.Indb
«Hun så veldig fl ink ut... Ansette henne?» 'SFNHBOHTSJL)3 XXXBTTFTTJPOP IT FINANS MARKED ENGINEERING Ring UpSource på 55 70 70 25 for en profesjonell vurdering. A-AVIS FOTO: ANITA MYKLEMYR ANITA FOTO: 14. MARS 2008 NR. 11 – 21. ÅRGANG – LØSSALG KR 25 Advokat Erling Grimstad: Kjerstin Fyllingen, –NAIV STATLIG TrygVesta: HOLDNING TIL HOPPET I LEDERROLLEN ETIKK SIDE 38 SIDE 32 FOTO: INGAR HAUG STEINHOLT/SCANPIXSTEINHOLT/SCANPIX Lær deg å like TIDS- Banker OPTIMISTEN SIDE 24–26 børsgutta FOTO: STIAN LYSBERG SOLUM/SCANPIX i ledelse Christian Rynning- Ukeavisen Ledelse har vurdert nærmere Tønnesen, Norske Skog: 40 toppsjefer. Etter å ha snakket med Papirtigeren 15 personer om Åse Aulie Michelets SIDE 4 sterke og svake sider som leder, kan vi GOD PÅSKE! konstatere at hun er noe av det beste Neste utgavegave av som kan oppdrives av sjefer her til lands. Ukeavisenn På bunnen av toppsjef-lista ligger sjefen Ledelse hennes, John Fredriksen. SIDE 15–20 kommer 28. mars. LEDERVERKTØY: SLIK BEHANDLER DU JOBBSØKERE SIDE 28 Åpen kontorløsning kan redusere kostnader og øke kompetansen i virksomheten! Mouversi har lang erfaring med gjennomføring av prosjekter ved etablering av åpen kontorløsning. Riktig fysisk utforming av arealet - Etablering av team - Økt medarbeiderkompetanse — Fornøyde kunder Utprøvd metode og gode resultater kan dokumenteres. Kontakt oss for rådgiving og prosessgjennomføring. Mouversi betyr å flytte på seg, både fysisk og samtidig flytte grenser for egne prestasjoner. mouversi flytter mennesker og virksomheter Fjordgt 9, 7011 Trondheim, Mobil: 93201177, Mail: [email protected] www.aapenkontorlosning.no 2 NR. 11 – FREDAG 14. MARS 2008 – UKEAVISEN LEDELSE LEDERPLASS FOTO: SVEIN ERIK FURULUND/SCANPIX Tilliten i markedet til Rynning-Tønnesen har fått et knekk, men det er for tidlig å regne med at han ikke får fortsette som konsernsjef i Norske Skog. -

Nycomed Joins List of Private Companies up for Sale
March 24, 2009 Nycomed joins list of private companies up for sale Amy Brown Reports claiming that Nycomed is seeking a buyer adds the Swiss-based specialty pharma company to the growing list of significantly sized private healthcare companies that are supposedly on the “for sale” list. The news followed claims last week that closely-held US dermatology group, Stiefel Laboratories, was seeking buyers, in a deal that could be worth $3bn. Last year, CLS bid $1.3bn for Talecris Biotherapeutics, another substantial private company take out should it complete. What all three have in common are private equity stake holders. The proximity of three big private company sales could be a coincidence. However, with many investors keen to shore up cash balances, the pressure to exit past investments is probably as intense as it has ever been. In August last year Talecris was sold by its private equity owners, Cerberus Capital and Ampersand Ventures, to CSL following an aborted IPO attempt the previous year. Its private equity owners were clearly very keen for an exit. A similar situation could be emerging with Nycomed, which is owned by four financial institutions. Unlike Talecris and Nycomed, Stiefel is still majority owned by the founding family; Blackstone owns a minority stake following a $500m investment in 2007. Credit crunch There are also privately owned generics groups currently on the block, although for different reasons. Actavis from Iceland and Germany’s Ratiopharm are up for sale through necessity rather than choice; their billionaire businessmen owners, crippled by the credit crunch, need to pay down debt. -

Pharma Congress Production & Technology 2014
PHARMA CONGRESS Production & Technology network. experience. benefi t. 14 DÜSSELDORF, 25 - 26 MARCH 2014 With more than 60 Speakers... Regulatory Presentations from: Scott Aldrich, USA Member of the 2010-2015 USP Dosage Forms Expert Committee Dr Ajaz S. Hussain, USA Former FDA Deputy Director Klaus Feuerhelm, Regierungspräsidium Tübingen GMP Inspektor Dr Jean-Denis Mallet Former Head of the Pharmaceutical Inspection Dpt. AFSSAPS Dr Daniel Müller, Regierungspräsidium Tübingen GMP Inspector Dr Stephen Langille, USA (invited) FDA Industry Case Studies from e.g.: Dr Friedrich Haefele, Boehringer Ingelheim Vice President BP Fill & Finish Germany Dr Helmut Gaus, Rentschler Biotechnologie Vice President Quality Control Dr Sanjit Singh Lamba, Eisai India Managing Director of Eisai Knowledge Centre, Head – Global Procurement Strategy ECA Conference Dr Barthold Piening, Takeda Pharmaceuticals Current Aseptic Technologies Head of Global Operations ECA Conference Polymer-based Philip Schneider, F. Hoffmann-La Roche Prefi lled Syringes Head of Sterile Drug Product Manufacturing Kaiseraugst ECA Conference Isolator Technology Patrick Vanhecke, GlaxoSmithKline Senior Manager at GlaxoSmithKline Biologicals ECA Conference Single-Use Disposables Dr Helmut Vigenschow, TEVA – Merckle Head of Quality Assurance in Germany ECA Conference Manufacture of Oral Solid Dosage Forms Jörg Zimmermann, Vetter Pharma-Fertigung Director Process Development and ECA Conference Implementation Particles in Parenterals Image: Copyright Lonza Image: Copyright PHARMA CONGRESS Production & Technology 14 DÜSSELDORF, 25 - 26 MARCH 2014 The Steering For the Pharma Congress 2014 Steering Committee we were able to win leading experts from Committee the pharmaceutical industry with extended knowlegde in production and technology. Their support allowed us to develop a programme that is even closer to what you're dealing with in your daily routine. -
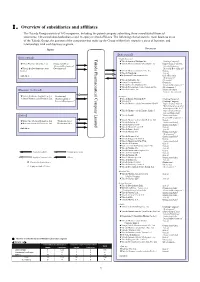
Ⅰ.Overview of Subsidiaries and Affiliates
Ⅰ.Overview of subsidiaries and affiliates The Takeda Group consists of 165 companies, including the parent company submitting these consolidated financial statements, 148 consolidated subsidiaries and 16 equity method affiliates. The following chart shows the main business areas of the Takeda Group, the position of the companies that make up the Group within their respective areas of business, and relationships with each business segment. Japan Overseas 【Ethical drugs】 【Ethical drugs】 Takeda Pharmaceutical Limited Pharmaceutical Company Takeda <North America> ★Takeda America Holdings, Inc. 《Holding Company》 ★Nihon Pharmaceutical Co., Ltd. 《Production/Sales/ ★Takeda Pharmaceuticals International, Inc. 《Supervision of the U.S. Research/Development》 Sales & Marketing/ ★Takeda Bio Development Center 《Development》 Research/ Development》 Limited ★Takeda Pharmaceuticals U.S.A., Inc. 《Sales》 ★Takeda Canada Inc. 《Sales》 And others ★Millennium Pharmaceuticals, Inc. 《Sales/Research/ Development》 ★Takeda California, Inc. 《Research》 ★Envoy Therapeutics, Inc. 《Research》 ★Takeda Vaccine (Montana), Inc. 《Research/Development 》 ★Takeda Development Center Americas, Inc. 《Development 》 【Consumer Healthcare】 ★Takeda Ventures, Inc. 《Research-related Venture Investment 》 ★Takeda Healthcare Products Co., Ltd. 《Production》 <Europe> ◎Amato Pharmaceutical Products, Ltd. 《Production/Sales/ ★Takeda Europe Holdings B.V. 《Holding Company》 Research/Development》 ★Takeda A/S 《Holding Company》 ★Takeda Pharmaceuticals International GmbH 《Supervision of Sales & Marketing for areas other than Japan and the U.S.》 ★Takeda Pharmaceuticals Europe, Limited 《Supervision of Europe Sales & Marketing》 【Others】 ★Takeda GmbH 《Production/Sales/ Research/Development 》 ★Takeda Pharma Vertrieb GmbH & Co. KG 《 》 ★Wako Pure Chemical Industries, Ltd. 《Production/Sales》 Sales ★Takeda Italia S.p.A. 《Production/Sales》 ★Mizusawa Industrial Chemicals, Ltd. 《Production/Sales》 ★Takeda Austria GmbH 《Production/Sales 》 ★Takeda Pharma Ges.m.b.H 《Sales 》 And others ★Takeda France S.A.S. -

PACIRA PHARMACEUTICALS, INC. (Exact Name of Registrant As Specified in Its Charter)
Table of Contents UNITED STATES SECURITIES AND EXCHANGE COMMISSION Washington, D.C. 20549 FORM 10-K (Mark One) x ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 For the fiscal year ended: December 31, 2010 Or ¨ TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 For the transition period from to Commission file number: 001-35060 PACIRA PHARMACEUTICALS, INC. (Exact Name of Registrant as Specified in Its Charter) Delaware 51-0619477 (State or Other Jurisdiction of (I.R.S. Employer Incorporation or Organization) Identification No.) 5 Sylvan Way, Suite 125 Parsippany, New Jersey 070504 (Address of Principal Executive Offices) (zip code) Registrant’s telephone number, including area code (973) 254-3560 Securities registered pursuant to Section 12(b) of the Act: Title of each class Name of each exchange on which registered Common Stock, $0.001 par value The NASDAQ Global Market Securities registered pursuant to Section 12(g) of the Act: None Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ¨ No x Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes ¨ No x Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. -

Corporate Profile 2017
CORPORATE PROFILE 2017 Takeda Pharmaceutical Company Limited CONTENTS 01 — OUR VALUES Who We Are 6 Our Heritage, Our Future 8 02 — OUR PEOPLE A Message from our CEO 16 Corporate Governance 18 Board of Directors 19 Takeda Executive Team 20 Diverse Perspectives 22 Talent Development 24 03 — RESEARCH & DEVELOPMENT Our Research 28 Takeda's Shonan Research Center is located between Kamakura and Fujisawa, about 50km south of Tokyo. The campus also Partnerships and Collaborations 30 includes a day-care center for employees children, and is surrounded by a rich variety of trees and wildlife. It is also home to the Shonan Health Innovation Park, which we aspire to become 04 — BUSINESS PERFORMANCE FY2016 Underlying Growth 34 the leading hub for Japan’s biopharmaceutical ecosystem. Takeda's Growth Drivers 35 Corporate Social Responsibility (CSR) 36 TAKEDA.COM Takeda's Global CSR Program 38 Published July 2017 Takeda Pharmaceutical Company Limited Access to Medicines 40 01 VALUES Takeda-ism and Our Priorities Originally built in 1932 as a residence for Chobei VI, the Kansokyo Takeda Historical Museum is located 7km east of Kobe, Japan. While externally it was modeled on the English Tudor style, its core structure was built of wood in a unique Japanese style, giving it both strength and the flexibility to withstand earthquakes. 4 TAKEDA CORPORATE PROFILE 2017 OUR Values 5 WHO WE ARE Takeda is a patient-focused, innovation driven YEARS global pharmaceutical company that builds on a distinguished236-year history. Our mission is to strive towards better health and a brighter future for people worldwide through leading innovation in medicine. -

Annual Report 2011 Annual Report
ORKLA A NNU AL R EP OR T / 2011 Annual Report 2011 IC ECOL D AB R E Orkla ASA Office address: Tel: +47 22 54 40 00 www.orkla.com O L N P.O. Box 423 Skøyen Karenslyst allé 6 Fax: +47 22 54 44 90 [email protected] NO-0213 Oslo, Norway NO-0278 Oslo, Norway Enterprise number: NO 910 747 711 Contents Message from the CEO 4 The Board of Directors of Orkla ASA 6 Report of the Board of Directors 8 The Group Executive Board 22 Corporate Governance 23 Annual Financial Statements 2011 32 Notes to the Financial Statements 37 Financial Statements for Orkla ASA 81 Historical key figures 90 Group Directory 91 Organisation 95 Governing bodies and elected representatives 96 Financial calendar for Orkla ASA DATE EVENT 19.04.2012 Annual General Meeting 20.04.2012 Share traded ex.-dividend* 03.05.2012 Dividend payment* Front page: The figures are a selection from the ornamental work of art unveiled at Orkla’s head office in 2011. The artist, Benjamin Bergman, has made a thorough study of 03.05.2012 1st quarter 2012 Orkla’s history. The work spans all six floors of the building, and consists of 18 levels and 357 figures, one from each year since Orkla was founded. 20.07.2012 2nd quarter 2012 31.10.2012 3rd quarter 2012 *Subject to the approval of the proposed dividend at the General Meeting. Design and production Cox Design, Oslo | Photo: Guri Dahl/tinagent.no | Photo above: Werner Anderson, Cox | Print: Aktiv Trykk AS Orkla_aars_eng_omslag_orig_1303.indd 3 13.03.12 18.24 / The Orkla Group / Orkla Annual Report 2011 The Orkla Group Orkla operates in the branded consumer goods, aluminium solutions, renewable energy, biobased chemicals and financial investment sectors. -

Relief Therapeutics Holding AG Switzerland | Biotechnology
Relief Therapeutics Holding AG Switzerland | Biotechnology Initiation of Coverage Addressing diabetes and respiratory disease 26 September 2016 Relief Therapeutics Holding AG (‘Relief Therapeutics’ or ‘Relief’ or ‘the company’) is a Switzerland-based biotechnology company developing a rich pipeline of candidates, primarily aimed at diabetic complications and Company Data respiratory diseases. The company is currently developing two drug candidates; Aviptadil, set to enter a pivotal Phase III trial in sarcoidosis, Price: CHF 0.03 and Atexakin alfa, aimed at peripheral neuropathies, which is slated to Market Cap: CHF58m enter Phase II testing in patients with diabetic neuropathy. Both studies Free Float (%): 12.8% could start in the first half of 2017. Atexakin alfa is the subject of a worldwide exclusive license from Merck KGaA and was originally developed Nr. Of shares: 1'946'627'375 by Serono S.A. Avg. traded vol. (1 year): 445'727 Attractive potential in underserved markets Bloomberg: RLF SW The company is developing drug candidates to address markets in Reuters: RLFB.S which there currently are no approved effective therapies. While ISIN: CH0100191136 Aviptadil targets orphan disease indications, including sarcoidosis and Source: SIX Swiss Exchange and Bloomberg acute lung injury (ALI), Atexakin alfa (recombinant human interleukin- 6, or rhIL-6) targets larger markets, including peripheral diabetic neuropathy (PDN) and chemotherapy-induced peripheral neuropathy (CIPN). Given the benign safety profile of Aviptadil and the lack of existing treatments for sarcoidosis, we anticipate higher penetration rates and pricing power in future for Aviptadil targeted for sarcoidosis. Atexakin alfa, on the other hand, could constitute the basis for a new Share Price Development class of neuropathy-targeting anti-nociceptive (pain-suppressing) drugs that may treat both the pain-related and non-pain related symptoms due to diabetes-induced neuropathic complications. -

FOIA Request & Response
June 22, 2018 Dear SEC FOIA Office: RECEIVE!:> I am requesting a copy of JUN 2? 2018 Exhibit 10.3 Medicines Co/Ma Form 10-Q dated 05/15/2002. Office of I am willing to pay up to $61.00. F'()l 11 ~ •• - ~ - ~ Thank you, Diane Martin AUS Consultants Inc. 155 Gaither Dr, Suite A Mt. Laurel NJ08054 856.234.9200 19 UNITED STATES SECURITIES AND EXCHANGE COMMISSION STATION PLACE 100 F STREET, NE WASHINGTON, DC 20549-2465 Office of FOIA Services July 6, 2018 Ms. Diane Martin AUS Consultants, Inc. 155 Gaither Dr., Suite A Mt. Laurel, NJ 08054 RE: Freedom of Information Act (FOIA), 5 U.S.C. § 552 Request No. 18-04916-E Dear Ms. Martin: This letter is in response to your request, dated and received in this office on June 22, 2018, for Exhibit 10.3 Medicines Co. Ma Form 10-Q, dated May 15, 2002. The search for responsive records has resulted in the retrieval of 43 pages of records that may be responsive to your request. They are being provided to you with this letter. If you have any questions, please contact me at [email protected] or (202) 551-8186. You may also contact me at [email protected] or (202) 551-7900. You also have the right to seek assistance from Lizzette Katilius as a FOIA Public Liaison or contact the Office of Government Information Services (OGIS) for dispute resolution services. OGIS can be reached at 1-877- 684-6448 or Archives.gov or via e-mail at [email protected].