Radiation-Induced Bystander Effects, Carcinogenesis and Models
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Testing the Stand-Alone Microbeam at Columbia University G
Radiation Protection Dosimetry Advance Access published December 21, 2006 Radiation Protection Dosimetry (2006), 1 of 5 doi:10.1093/rpd/ncl454 TESTING THE STAND-ALONE MICROBEAM AT COLUMBIA UNIVERSITY G. GartyÃ, G. J. Ross, A. W. Bigelow, G. Randers-Pehrson and D. J. Brenner Columbia University, Radiological Research Accelerator Facility, 136 S. Broadway, Irvington, NY 10533, USA The stand-alone microbeam at Columbia University presents a novel approach to biological microbeam irradiation studies. Foregoing a conventional accelerator as a source of energetic ions, a small, high-specific-activity, alpha emitter is used. Alpha particles emitted from this source are focused using a compound magnetic lens consisting of 24 permanent magnets arranged in two quadrupole triplets. Using a ‘home made’ 6.5 mCi polonium source, a 1 alpha particle s–1,10lm diameter microbeam can, in principle, be realised. As the alpha source energy is constant, once the microbeam has been set up, no further adjustments are necessary apart from a periodic replacement of the source. The use of permanent magnets eliminates the need for bulky power supplies and cooling systems required by other types of ion lenses and greatly simplifies operation. It also makes the microbeam simple and cheap enough to be realised in any large lab. The Microbeam design as well as first tests of its performance, using an accelerator-based beam are presented here. INTRODUCTION AND OVERALL DESIGN the lenses. In addition to providing a secondary user facility at RARAF, the design is simple and Columbia University’s Radiological Research Accel- inexpensive enough that the SAM can be reproduced erator Facility (RARAF) currently offers its users in any large radiation biology laboratory. -

A New Application of Microbeam Radiation Therapy (MRT) on the Treatment of Epilepsy and Brain Disorders
A new application of microbeam radiation therapy (MRT) on the treatment of epilepsy and brain disorders. Erminia Fardone To cite this version: Erminia Fardone. A new application of microbeam radiation therapy (MRT) on the treatment of epilepsy and brain disorders.. Human health and pathology. Université de Grenoble, 2013. English. NNT : 2013GRENV060. tel-01552806 HAL Id: tel-01552806 https://tel.archives-ouvertes.fr/tel-01552806 Submitted on 3 Jul 2017 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. THÈSE Pour obtenir le grade de DOCTEUR DE L’UNIVERSITÉ DE GRENOBLE Spécialité : Neurosciences - Neurobiologie Arrêté ministériel : 7 août 2006 Présentée par Erminia Fardone Thèse dirigée par Serge PEREZ codirigée par Alberto BRAVIN et Pantaleo ROMANELLI préparée au sein de l’ Installation européenne de rayonnement synchrotron (ESRF) dans l'École Doctorale Chimie et Sciences du Vivant A new application of microbeam radiation therapy (MRT) on the treatment of epilepsy and brain disorders Thèse soutenue publiquement le Vendredi 29 Novembre 2013, devant le jury composé de : Prof Alain Buisson (Président) Prof Université Joseph Fourier, Grenoble, France Prof Paola Coan (Examinateur) Prof Ludwig Maximilians University, Munich, Germany Prof Guido Cavaletti (Rapporteur) Prof University of Milan-Bicocca, Italy Prof Elisabeth Schültke (Rapporteur) MD, Dr Freiburg University Medical Center, Freiburg, Germany Dr Serge Perez (Directeur de Thèse) D.R. -

Animal Models in Microbeam Radiation Therapy: a Scoping Review
cancers Review Animal Models in Microbeam Radiation Therapy: A Scoping Review Cristian Fernandez-Palomo 1 , Jennifer Fazzari 1, Verdiana Trappetti 1 , Lloyd Smyth 2 , Heidrun Janka 3, Jean Laissue 1 and Valentin Djonov 1,* 1 Institute of Anatomy, University of Bern, 3012 Bern, Switzerland; [email protected] (C.F.-P.); [email protected] (J.F.); [email protected] (V.T.); [email protected] (J.L.) 2 Department of Obstetrics & Gynaecology, University of Melbourne, 3057 Parkville, Australia; [email protected] 3 Medical Library, University Library Bern, University of Bern, 3012 Bern, Switzerland; [email protected] * Correspondence: [email protected]; Tel.: +41-31-631-8432 Received: 24 January 2020; Accepted: 21 February 2020; Published: 25 February 2020 Abstract: Background: Microbeam Radiation Therapy (MRT) is an innovative approach in radiation oncology where a collimator subdivides the homogeneous radiation field into an array of co-planar, high-dose beams which are tens of micrometres wide and separated by a few hundred micrometres. Objective: This scoping review was conducted to map the available evidence and provide a comprehensive overview of the similarities, differences, and outcomes of all experiments that have employed animal models in MRT. Methods: We considered articles that employed animal models for the purpose of studying the effects of MRT. We searched in seven databases for published and unpublished literature. Two independent reviewers screened citations for inclusion. Data extraction was done by three reviewers. Results: After screening 5688 citations and 159 full-text papers, 95 articles were included, of which 72 were experimental articles. -

The Oncogenic Transforming Potential of the Passage of Single Particles
Proc. Natl. Acad. Sci. USA Vol. 96, pp. 19–22, January 1999 Applied Biological Sciences The oncogenic transforming potential of the passage of single a particles through mammalian cell nuclei RICHARD C. MILLER,GERHARD RANDERS-PEHRSON,CHARLES R. GEARD,ERIC J. HALL, AND DAVID J. BRENNER* Center for Radiological Research, Columbia University, 630 West 168th Street, New York, NY 10032 Communicated by Richard B. Setlow, Brookhaven National Laboratory, Upton, NY, November 3, 1998 (received for review June 2, 1998) ABSTRACT Domestic, low-level exposure to radon gas is cell at risk will be traversed by more than one a particle in a considered a major environmental lung-cancer hazard involv- lifetime (1). ing DNA damage to bronchial cells by a particles from radon Even in the laboratory, there has until now been no direct progeny. At domestic exposure levels, the relevant bronchial way of measuring the oncogenic transforming effects of exactly cells are very rarely traversed by more than one a particle, one a particle without the confounding effects of a significant whereas at higher radon levels—at which epidemiological fraction of exposed cells being subject to multiple a-particle studies in uranium miners allow lung-cancer risks to be traversals, and this has led to significant uncertainty in low- quantified with reasonable precision—these bronchial cells dose radon risk estimates (2). are frequently exposed to multiple a-particle traversals. Mea- In recent years, several groups have developed charged- suring the oncogenic transforming effects of exactly one a particle microbeams, in which cells on a dish are individually particle without the confounding effects of multiple traversals irradiated by a predefined exact number of a particles, thus has hitherto been unfeasible, resulting in uncertainty in allowing the effects of exactly one (or more) a particle extrapolations of risk from high to domestic radon levels. -

Induction of a Bystander Mutagenic Effect of Alpha Particles in Mammalian Cells
Induction of a bystander mutagenic effect of alpha particles in mammalian cells Hongning Zhou*, Gerhard Randers-Pehrson*, Charles A. Waldren†, Diane Vannais†, Eric J. Hall*, and Tom K. Hei*‡§ *Center for Radiological Research, College of Physicians and Surgeons, and ‡Environmental Health Sciences, School of Public Health, Columbia University, New York, NY 10032; and †Department of Radiological Health Sciences, Colorado State University, Fort Collins, CO 80523 Edited by Richard B. Setlow, Brookhaven National Laboratory, Upton, NY, and approved December 10, 1999 (received for review October 1, 1999) Ever since the discovery of X-rays was made by Ro¨ntgen more than effect would require an area 350 times the typical size of a CHO a hundred years ago, it has always been accepted that the dele- nucleus (9). The additional responding cells that received no terious effects of ionizing radiation such as mutation and carcino- irradiation were ‘‘bystanders’’ of either directly hit cells or genesis are attributable mainly to direct damage to DNA. Although resulted from agents released from the irradiated medium (8, evidence based on microdosimetric estimation in support of a 10). Subsequent studies suggested that reactive oxygen species bystander effect appears to be consistent, direct proof of such might contribute to the induction of SCE among the bystander extranuclear͞extracellular effects are limited. Using a precision cells (11). Enhanced expression of the p53 tumor suppressor charged particle microbeam, we show here that irradiation of 20% gene in bystander cells has also been reported in immortalized of randomly selected AL cells with 20 alpha particles each results in rat lung epithelial cells and human diploid fibroblast cells a mutant fraction that is 3-fold higher than expected, assuming no irradiated with alpha particles (12, 13). -

Biological Measure of DNA Damage After Single-Ion Microbeam
Biological measure of DNA damage after single-ion microbeam irradiation and Monte Carlo simulations Geraldine Gonon, Carmen Villagrasa, Pascale Voisin, Sylvain Meylan, Mohamedamine Benadjaoud, Nicolas Tang, Frank Langner, Hans Rabus, Joan Francesc Barquinero, Ulrich Giesen, et al. To cite this version: Geraldine Gonon, Carmen Villagrasa, Pascale Voisin, Sylvain Meylan, Mohamedamine Benadjaoud, et al.. Biological measure of DNA damage after single-ion microbeam irradiation and Monte Carlo sim- ulations. 16th International Congress of Radiation Research ICRR 2019, Aug 2019, MANCHESTER, France. 2019. hal-02635559 HAL Id: hal-02635559 https://hal.archives-ouvertes.fr/hal-02635559 Submitted on 27 May 2020 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Copyright N° 404 Biological measure of DNA damage after single-ion microbeam irradiation and Monte Carlo simulations Géraldine Gonon1,Carmen Villagrasa1, Pascale Voisin1, Sylvain Meylan1, Mohamed Amine Benadjaoud1, Nicolas Tang1, Frank Langner2, Hans Rabus2, Joan Francesc Barquinero3, Ulrich Giesen2, Gaëtan -

Collimated Microbeam Reveals That the Proportion of Non-Damaged
biology Article Collimated Microbeam Reveals that the Proportion of Non-Damaged Cells in Irradiated Blastoderm Determines the Success of Development in Medaka (Oryzias latipes) Embryos Takako Yasuda 1,*, Tomoo Funayama 2 , Kento Nagata 1, Duolin Li 1, Takuya Endo 1, Qihui Jia 1, Michiyo Suzuki 2 , Yuji Ishikawa 3, Hiroshi Mitani 1 and Shoji Oda 1 1 Department of Integrated Biosciences, Graduate School of Frontier Sciences, The University of Tokyo, Chiba 277-8562, Japan; [email protected] (K.N.); [email protected] (D.L.); [email protected] (T.E.); [email protected] (Q.J.); [email protected] (H.M.); [email protected] (S.O.) 2 Takasaki Advanced Radiation Research Institute, Quantum Beam Science Research Directorate, National Institutes for Quantum and Radiological Science and Technology (QST), Gunma 370-1292, Japan; [email protected] (T.F.); [email protected] (M.S.) 3 National Institute of Radiological Sciences, Quantum Medical Science Directorate, National Institutes for Quantum and Radiological Science and Technology (QST), Chiba 263-8555, Japan; [email protected] * Correspondence: [email protected]; Tel.: +81-4-7136-3663; Fax: +81-4-7136-3669 Received: 2 October 2020; Accepted: 29 October 2020; Published: 5 December 2020 Simple Summary: Studies on teratogenesis in mammals have revealed that exposure to ionizing radiation (IR) during the pre-implantation period induces a high frequency of lethality instead of teratogenesis. Here, to elucidate the IR-induced disturbance of embryonic development when IR exposure occurs during the pre-implantation period, we utilized medaka as a vertebrate model for clear observation of developmental process for its transparency. -

Microbeam Advanced Example Located in $G4INSTALL/Examples/Advanced Contact : Sébastien Incerti ([email protected])
microbeam Advanced example located in $G4INSTALL/examples/advanced Contact : Sébastien Incerti ([email protected]) Overview The microbeam example simulates the microbeam cellular irradiation beam line installed on the AIFIRA electrostatic accelerator facility located at CENBG, Bordeaux-Gradignan, France. This setup is mainly used to investigate the effects of low dose irradiation on living cells. The microbeam line allows irradiation of individual biological living cells in culture medium in single ion mode, with an exact control of the delivered dose to a particular cell among the cell population. This Geant4 example simulates the microbeam line in a configuration of irradiation with 3 MeV incident alpha particles and allows the calculation of the dose deposited by the incident particles in the cell cytoplasm and in the cell nucleus, which are inaccessible by experimental measurements. For the first time in Monte Carlo microdosimetry, the simulation includes a realistic cell phantom obtained from confocal microscopy and from ion beam anlysis techniques. For more information on this irradiation facility, please visit : http://www.cenbg.in2p3.fr Schematic view of the CENBG five AIFIRA beam lines View of the CENBG microbeam line microbeam Description The beam is emitted just before the 10° switching magnet taking into account experimental beam parameters measurements ; the main elements simulated are : • A switching dipole magnet with fringing field, to deflect by 10° the 3 MeV alpha beam generated by the electrostatic accelerator into the microbeam line, oriented at 10 degrees from the main beam direction; • A circular object collimator, defining the incident beam size at the microbeam line entrance; the collimator has been simulated from realistic electron microscopy images; • A quadrupole based magnetic symmetric focusing system allowing equal transverse demagnifications of 10. -

Microbeam Studies of the Sensitivity of Structures Within Living Cells
Scanning Microscopy Volume 6 Number 1 Article 13 12-30-1991 Microbeam Studies of the Sensitivity of Structures within Living Cells L. A. Braby Pacific Northwest Laboratory Follow this and additional works at: https://digitalcommons.usu.edu/microscopy Part of the Biology Commons Recommended Citation Braby, L. A. (1991) "Microbeam Studies of the Sensitivity of Structures within Living Cells," Scanning Microscopy: Vol. 6 : No. 1 , Article 13. Available at: https://digitalcommons.usu.edu/microscopy/vol6/iss1/13 This Article is brought to you for free and open access by the Western Dairy Center at DigitalCommons@USU. It has been accepted for inclusion in Scanning Microscopy by an authorized administrator of DigitalCommons@USU. For more information, please contact [email protected]. Scanning Microscopy, Vol. 6, No. 1, 1992 (Pages 167-175) 0891-7035/92$5 .00+ .00 Scanning Microscopy International, Chicago (AMF O'Hare), IL 60666 USA MICROBEAMSTUDIES OF THESENSITIVITY OF STRUCTURESWITHIN LIVING CELLS L.A. Braby* Battelle, Pacific Northwest Laboratory Richland WA.99352 (Received for publication May 4, 1991, and in revised form December 30, 1991) ABSTRACT INTRODUCTION Determining the biological effects of low doses A new generation of charged-particle microbeam of radiation with high linear energy transfer (LET) irradiation systems is being installed at several is complicated by the stochastic nature of charged laboratories around the world. These systems have particle interactions. Populations of cells been designed to answer some fundamental questions exposed to very low radiation doses contain a few about the hazards of low doses of ionizing radia cells which have been hit by a charged particle, tion. -

Alpha Emitters for Radiotherapy: Basic Radiochemistry to Clinical Studies Part 1
Journal of Nuclear Medicine, published on March 15, 2018 as doi:10.2967/jnumed.116.186338 Alpha Emitters for Radiotherapy: Basic Radiochemistry to Clinical Studies _ Part 1 Running title: Alpha Emitters for Radiotherapy Sophie Poty1, Lynn C. Francesconi2,3, Michael R. McDevitt1,4, Michael J. Morris,5 Jason S. Lewis1,6 1Department of Radiology and the Program in Molecular Pharmacology, Memorial Sloan Kettering Cancer Center, New York, NY, USA 2Department of Chemistry, Hunter College, New York, NY, USA 3The Graduate Center of the City University of New York, New York, NY, USA 4Departments of Radiology, Weill Cornell Medical College, New York, NY, USA 5Department of Medicine, Memorial Sloan Kettering Cancer Center, New York, NY, USA 6Departments of Radiology and Pharmacology, Weill Cornell Medical College, New York, NY, USA Correspondence: Jason S. Lewis, PhD, 1275 York Avenue, New York, NY 10065, USA. Phone: 646-888-3038, FAX: 646-422-0408, Email: [email protected] First author: Sophie Poty, PhD, 1275 York Avenue, New York, NY 10065, USA. Phone: 646- 888-3080, FAX: 646-422-0408, Email: [email protected] Word count: 5317 Disclaimer: The authors have nothing to disclose. Financial support: The authors gratefully acknowledge the Radiochemistry and Molecular Imaging Probe core, which was supported in part by the NIH/NCI Cancer Center Support Grant P30 CA008748. We gratefully acknowledge Mr. William H. and Mrs. Alice Goodwin and the Commonwealth Foundation for Cancer Research and The Center for Experimental Therapeutics of Memorial Sloan Kettering Cancer Center (JSL) and the fellowship from the François Wallace Monahan Fellowship from the JLM Benevolent Fund (SP). -

Field Size Effects on DNA Damage and Proliferation in Normal Human Cell
www.nature.com/scientificreports OPEN Field size efects on DNA damage and proliferation in normal human cell populations irradiated with X‑ray microbeams Mitsuaki Ojima1*, Atsushi Ito2, Noriko Usami3, Maki Ohara3, Keiji Suzuki4 & Michiaki Kai1 To clarify the health risks of internal radiation exposure, it is important to investigate the radiological efects of local exposure at cell levels from radioactive materials taken up by organs. Focusing on the response of cell populations post‑irradiation, X‑ray microbeams are very efective at reproducing the efects of local exposure within an internal exposure in vitro. The present study aims to clarify the efects of local exposure by investigating the response of normal human cell (MRC‑5) populations irradiated with X‑ray microbeams of diferent beam sizes to DNA damage. The populations of MRC‑5 were locally irradiated with X‑ray microbeams of 1 Gy at 0.02–1.89 mm2 feld sizes, and analyzed whether the number of 53BP1 foci as DSB (DNA double strand break) per cell changed with the feld size. We found that even at the same dose, the number of DSB per cell increased depending on the X‑irradiated feld size on the cell population. This result indicated that DNA damage repair of X‑irradiated cells might be enhanced in small size felds surrounded by non‑irradiated cells. This study suggests that X‑irradiated cells received some signal (a rescue signal) from surrounding non‑irradiated cells may be involved in the response of cell populations post‑irradiation. Insoluble radioactive cesium was released into the atmosphere due to the Fukushima Daiichi Nuclear Power Plant accident that occurred afer the Great East Japan Earthquake on March 11, 2011 1. -
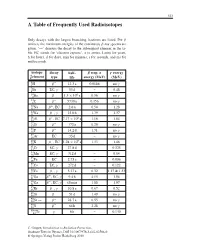
A Table of Frequently Used Radioisotopes
323 A Table of Frequently Used Radioisotopes Only decays with the largest branching fractions are listed. For β emitters the maximum energies of the continuous β-ray spectra are given. ‘→’ denotes the decay to the subsequent element in the ta- ble. EC stands for ‘electron capture’, a (= annus, Latin) for years, h for hours, d for days, min for minutes, s for seconds, and ms for milliseconds. isotope decay half- β resp. α γ energy A Z element type life energy (MeV) (MeV) 3 β− . γ 1H 12 3a 0.0186 no 7 γ 4Be EC, 53 d – 0.48 10 β− . × 6 γ 4Be 1 5 10 a 0.56 no 14 β− γ 6C 5730 a 0.156 no 22 β+ . 11Na ,EC 2 6a 0.54 1.28 24 β− γ . 11Na , 15 0h 1.39 1.37 26 β+ . × 5 13Al ,EC 7 17 10 a 1.16 1.84 32 β− γ 14Si 172 a 0.20 no 32 β− . γ 15P 14 2d 1.71 no 37 γ 18Ar EC 35 d – no 40 β− . × 9 19K ,EC 1 28 10 a 1.33 1.46 51 γ . 24Cr EC, 27 8d – 0.325 54 γ 25Mn EC, 312 d – 0.84 55 . 26Fe EC 2 73 a – 0.006 57 γ 27Co EC, 272 d – 0.122 60 β− γ . 27Co , 5 27 a 0.32 1.17 & 1.33 66 β+ γ . 31Ga , EC, 9 4h 4.15 1.04 68 β− γ 31Ga , EC, 68 min 1.88 1.07 85 β− γ .