Rapid Evolutionary Adaptation in Response to Selection on Quantitative Traits
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

An Introgression Analysis of Quantitative Trait Loci That Contribute to a Morphological Difference Between Drosophila Simulans and D
Copyight 0 1997 by the Genetics Societv of America An Introgression Analysis of Quantitative Trait Loci That Contribute to a Morphological Difference Between Drosophila simulans and D. mauritiana Cathy C. Laurie, John R. True, Jianjun Liu and John M. Mercer Department of Zoology, Duke University, Durham, North Carolina 27708 Manuscript received August 12, 1996 Accepted for publication October 29, 1996 ABSTRACT Drosophila simulans and D. maum'tiana differ markedly in morphology of the posterior lobe, a male- specific genitalic structure. Bothsize and shapeof the lobe can be quantifiedby a morphometric variable, PC1, derived from principal components and Fourier analyses. The genetic architectureof the species difference in PC1 was investigated previously by composite interval mapping, which revealed largely additive inheritance, with a minimum of eight quantitative trait loci (QTL) affecting the trait. This analysis was extended by introgression of marked segments of the maun'tiana third chromosome into a simulans background by repeated backcrossing. Thetwo types of experiment are consistentin suggesting that several QTL on the third chromosome may have effects in the range of 10-15% of the parental difference andthat all or nearly all QTL have effects in the same direction. Since the parental difference is large (30.4 environmental standard deviations), effects of this magnitude can produce alternative homozygotes with little overlap in phenotype. However, these estimates may not reflect the effects of individual loci, since each interval or introgressed segment may contain multiple QTL. The consistent direction of allelic effects suggests a history of directional selection on the posterior lobe. HE genetic analysis of a quantitative trait usually ual genes that affect continuously variable traits. -

Response to Selection in Finite Locus Models with Nonadditive Effects
Journal of Heredity, 2017, 318–327 doi:10.1093/jhered/esw123 Original Article Advance Access publication January 12, 2017 Original Article Response to Selection in Finite Locus Models with Nonadditive Effects Hadi Esfandyari, Mark Henryon, Peer Berg, Jørn Rind Thomasen, Downloaded from https://academic.oup.com/jhered/article/108/3/318/2895120 by guest on 23 September 2021 Piter Bijma, and Anders Christian Sørensen From the Center for Quantitative Genetics and Genomics, Department of Molecular Biology and Genetics, Aarhus University DK-8830 Tjele, Denmark (Esfandyari, Berg, Thomasen, and Sørensen); Seges, Danish Pig Research Centre, Copenhagen, Denmark (Henryon); School of Animal Biology, University of Western Australia, Crawley, Australia (Henryon); Nordic Genetic Resource Center, Ås, Norway (Berg); VikingGenetics, Assentoft, Denmark (Thomasen); and Animal Breeding and Genomics Centre, Wageningen University, Wageningen, The Netherlands (Bijma). Address correspondence to H. Esfandyari at the address above, or e-mail: hadi.esfandyari@ mbg.au.dk. Received May 18, 2016; First decision June 7, 2016; Accepted January 2, 2017. Corresponding Editor: C Scott Baker Abstract Under the finite-locus model in the absence of mutation, the additive genetic variation is expected to decrease when directional selection is acting on a population, according to quantitative-genetic theory. However, some theoretical studies of selection suggest that the level of additive variance can be sustained or even increased when nonadditive genetic effects are present. We tested the hypothesis that finite-locus models with both additive and nonadditive genetic effects maintain more additive genetic variance (VA) and realize larger medium- to long-term genetic gains than models with only additive effects when the trait under selection is subject to truncation selection. -

Microevolution and the Genetics of Populations Microevolution Refers to Varieties Within a Given Type
Chapter 8: Evolution Lesson 8.3: Microevolution and the Genetics of Populations Microevolution refers to varieties within a given type. Change happens within a group, but the descendant is clearly of the same type as the ancestor. This might better be called variation, or adaptation, but the changes are "horizontal" in effect, not "vertical." Such changes might be accomplished by "natural selection," in which a trait within the present variety is selected as the best for a given set of conditions, or accomplished by "artificial selection," such as when dog breeders produce a new breed of dog. Lesson Objectives ● Distinguish what is microevolution and how it affects changes in populations. ● Define gene pool, and explain how to calculate allele frequencies. ● State the Hardy-Weinberg theorem ● Identify the five forces of evolution. Vocabulary ● adaptive radiation ● gene pool ● migration ● allele frequency ● genetic drift ● mutation ● artificial selection ● Hardy-Weinberg theorem ● natural selection ● directional selection ● macroevolution ● population genetics ● disruptive selection ● microevolution ● stabilizing selection ● gene flow Introduction Darwin knew that heritable variations are needed for evolution to occur. However, he knew nothing about Mendel’s laws of genetics. Mendel’s laws were rediscovered in the early 1900s. Only then could scientists fully understand the process of evolution. Microevolution is how individual traits within a population change over time. In order for a population to change, some things must be assumed to be true. In other words, there must be some sort of process happening that causes microevolution. The five ways alleles within a population change over time are natural selection, migration (gene flow), mating, mutations, or genetic drift. -

Directional Selection and Variation in Finite Populations
Copyright 0 1987 by the Genetics Society of America Directional Selection and Variation in Finite Populations Peter D. Keightley and ‘William G. Hill Institute of Animal Genetics, University of Edinburgh, West Mains Road, Edinburgh EH9 3JN, Scotland Manuscript received April 12, 1987 Revised copy accepted July 16, 1987 ABSTRACT Predictions are made of the equilibrium genetic variances and responses in a metric trait under the joint effects of directional selection, mutation and linkage in a finite population. The “infinitesimal model“ is analyzed as the limiting case of many mutants of very small effect, otherwise Monte Carlo simulation is used. If the effects of mutant genes on the trait are symmetrically distributed and they are unlinked, the variance of mutant effects is not an important parameter. If the distribution is skewed, unless effects or the population size is small, the proportion of mutants that have increasing effect is the critical parameter. With linkage the distribution of genotypic values in the population becomes skewed downward and the equilibrium genetic variance and response are smaller as disequilibrium becomes important. Linkage effects are greater when the mutational variance is contributed by many genes of small effect than few of large effect, and are greater when the majority of mutants increase rather than decrease the trait because genes that are of large effect or are deleterious do not segregate for long. The most likely conditions for ‘‘Muller’s ratchet” are investi- gated. N recent years there has been much interest in the is attained more quickly in the presence of selection I production and maintenance of variation in pop- than in its absence, and is highly dependent on popu- ulations by mutation, stimulated by the presence of lation size. -

Selection Response in Finite Populations
Copyright 0 1996 by the Genetics Society of America Selection Responsein Finite Populations Ming Wei, Armando Caballero and William G. Hill Institute of Cell, Animal and Population Biology, University of Edinburgh, King’s Buildings, Edinburgh EH9 3JT, United Kingdom Manuscript received November 28, 1995 Accepted for publication September 5, 1996 ABSTRACT Formulae were derived to predict genetic response under various selection schemes assuming an infinitesimal model. Accountwas taken of genetic drift, gametic (linkage) disequilibrium (Bulmereffect), inbreeding depression, common environmental variance, and both initial segregating variance within families (a;wo)and mutational (&) variance. The cumulative response to selection until generation t(C8) can be approximated as where N, is the effective population size, = Ne& is the genetic variancewithin families at the steady state (or one-half the genic variance, which is unaffected by selection), and D is the inbreeding depression per unit of inbreeding. & is the selection response at generation 0 assuming preselection so that the linkage disequilibrium effect has stabilized. P is the derivative of the logarithm of the asymptotic response with respect to the logarithm of the within-family genetic variance, ie., their relative rate of change. & is the major determinant of the short term selection response, but aL, Ne and p are also important for the long term. A selection method of high accuracy using family information gives a small N, and will lead to a larger- response in the short term and a smaller response in the long term, utilizing mutation less efficiently. ENETIC response to one cycle of selection for a 1975) using all information available are formally the G quantitative character is solely a function of selec- methods to achieve the maximum response for one tion accuracy (the correlation between the selection generation of selection. -

The Qtn Program and the Alleles That Matter for Evolution: All That’S Gold Does Not Glitter
PERSPECTIVE doi:10.1111/j.1558-5646.2011.01486.x THE QTN PROGRAM AND THE ALLELES THAT MATTER FOR EVOLUTION: ALL THAT’S GOLD DOES NOT GLITTER Matthew V. Rockman1,2 1Department of Biology and Center for Genomics and Systems Biology, New York University, 12 Waverly Place, New York, NY 10003 2E-mail: [email protected] Received April 20, 2011 Accepted September 30, 2011 The search for the alleles that matter, the quantitative trait nucleotides (QTNs) that underlie heritable variation within populations and divergence among them, is a popular pursuit. But what is the question to which QTNs are the answer? Although their pursuit is often invoked as a means of addressing the molecular basis of phenotypic evolution or of estimating the roles of evolutionary forces, the QTNs that are accessible to experimentalists, QTNs of relatively large effect, may be uninformative about these issues if large-effect variants are unrepresentative of the alleles that matter. Although 20th century evolutionary biology generally viewed large-effect variants as atypical, the field has recently undergone a quiet realignment toward a view of readily discoverable large- effect alleles as the primary molecular substrates for evolution. I argue that neither theory nor data justify this realignment. Models and experimental findings covering broad swaths of evolutionary phenomena suggest that evolution often acts via large numbers of small-effect polygenes, individually undetectable. Moreover, these small-effect variants are different in kind, at the molecular level, from the large-effect alleles accessible to experimentalists. Although discoverable QTNs address some fundamental evolutionary questions, they are essentially misleading about many others. -

Patterns and Power of Phenotypic Selection in Nature
Articles Patterns and Power of Phenotypic Selection in Nature JOEL G. KINGSOLVER AND DAVID W. PFENNIG Phenotypic selection occurs when individuals with certain characteristics produce more surviving offspring than individuals with other characteristics. Although selection is regarded as the chief engine of evolutionary change, scientists have only recently begun to measure its action in the wild. These studies raise numerous questions: How strong is selection, and do different types of traits experience different patterns of selection? Is selection on traits that affect mating success as strong as selection on traits that affect survival? Does selection tend to favor larger body size, and, if so, what are its consequences? We explore these questions and discuss the pitfalls and future prospects of measuring selection in natural populations. Keywords: adaptive landscape, Cope’s rule, natural selection, rapid evolution, sexual selection henotypic selection occurs when individuals with selection on traits that affect survival stronger than on those Pdifferent characteristics (i.e., different phenotypes) that affect only mating success? In this article, we explore these differ in their survival, fecundity, or mating success. The idea and other questions about the patterns and power of phe- of phenotypic selection traces back to Darwin and Wallace notypic selection in nature. (1858), and selection is widely accepted as the primary cause of adaptive evolution within natural populations.Yet Darwin What is selection, and how does it work? never attempted to measure selection in nature, and in the Selection is the nonrandom differential survival or repro- century following the publication of On the Origin of Species duction of phenotypically different individuals. -

Alleles Versus Mutations: Understanding the Evolution of Genetic Architecture Requires a Molecular Perspective on Allelic Origins
Alleles versus mutations: Understanding the evolution of genetic architecture requires a molecular perspective on allelic origins By: David L. Remington Remington, D. L. (2015). Alleles versus mutations: understanding the evolution of genetic architecture requires a molecular perspective on allelic origins. Evolution: international journal of organic evolution. doi:10.1111/evo.12775 "This is the peer reviewed version of the following article: Remington, D. L. (2015). Alleles versus mutations: understanding the evolution of genetic architecture requires a molecular perspective on allelic origins. Evolution: international journal of organic evolution. doi:10.1111/evo.12775, which has been published in final form at http://dx.doi.org/10.1111/evo.12775. This article may be used for non-commercial purposes in accordance with Wiley Terms and Conditions for Self-Archiving." Abstract: Perspectives on the role of large-effect quantitative trait loci (QTL) in the evolution of complex traits have shifted back and forth over the past few decades. Different sets of studies have produced contradictory insights on the evolution of genetic architecture. I argue that much of the confusion results from a failure to distinguish mutational and allelic effects, a limitation of using the Fisherian model of adaptive evolution as the lens through which the evolution of adaptive variation is examined. A molecular-based perspective reveals that allelic differences can involve the cumulative effects of many mutations plus intragenic recombination, a model that is supported by extensive empirical evidence. I discuss how different selection regimes could produce very different architectures of allelic effects under a molecular-based model, which may explain conflicting insights on genetic architecture from studies of variation within populations versus between divergently selected populations. -
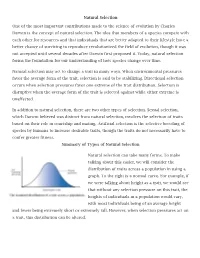
Natural Selection One of the Most Important Contributions Made to The
Natural Selection One of the most important contributions made to the science of evolution by Charles Darwin is the concept of natural selection. The idea that members of a species compete with each other for resources and that individuals that are better adapted to their lifestyle have a better chance of surviving to reproduce revolutionized the field of evolution, though it was not accepted until several decades after Darwin first proposed it. Today, natural selection forms the foundation for our understanding of how species change over time. Natural selection may act to change a trait in many ways. When environmental pressures favor the average form of the trait, selection is said to be stabilizing. Directional selection occurs when selection pressures favor one extreme of the trait distribution. Selection is disruptive when the average form of the trait is selected against while either extreme is unaffected. In addition to natural selection, there are two other types of selection. Sexual selection, which Darwin believed was distinct from natural selection, involves the selection of traits based on their role in courtship and mating. Artificial selection is the selective breeding of species by humans to increase desirable traits, though the traits do not necessarily have to confer greater fitness. Summary of Types of Natural Selection Natural selection can take many forms. To make talking about this easier, we will consider the distribution of traits across a population in using a graph. To the right is a normal curve. For example, if we were talking about height as a trait, we would see that without any selection pressure on this trait, the heights of individuals in a population would vary, with most individuals being of an average height and fewer being extremely short or extremely tall. -

100 Years of Quantitative Genetics Theory and Its Applications: Celebrating the Centenary of Fisher 1918
100 years of quantitative genetics theory and its applications: celebrating the centenary of Fisher 1918 Nick Barton Institute for Science and Technology, Klosterneuburg, Austria The infinitesimal model In his 1918 paper, Fisher introduced the analysis of variance, and reconciled Mendelian genetics with continuous biometric variation. He also showed that when variation is due to very many additive genetic loci, selection hardly changes the genetic variance. This has come to be known as the “infinitesimal model”, and is the foundation for practical animal breeding. Yet, organisms are plainly not simply the sum of their genes, and so it is not clear when the use of the infinitesimal model can be justified. I review the history of the infinitesimal model, and show how it can be extended to include dominance, epistasis, and linkage. Sharon Browning University of Washington, Seattle, Washington, USA Identity by descent and the correlation between distant relatives Identity by descent and the correlation between distant relatives Related individuals have correlated trait values because they share parts of their genomes identical by descent. Because the genome is inherited in large chunks, punctuated by crossovers, the identity by descent (IBD) occurs in long segments. IBD segments inherited from more distant ancestors tend to be shorter due to more opportunities for crossing over. So-called “unrelated” individuals are in fact distant relatives, and share short segments of IBD. I will describe the detection of segments of IBD in population samples using genome-wide genetic data, and the application of these segments to IBD mapping and to estimating heritability. Edward Buckler USDA-ARS, Ithaca, New York, USA How to get to Plant Breeding 4.0, given that Fisher was right? As global population continues to grow and standards of living rise, the world food supply needs to almost double protein production. -

Animal Breeding from Infinitesimal Model to Mas: the Case of a Backcross Design in Dairy Sheep (Sarda X Lacaune) and Its Possible Impact on Selection
MARKER ASSISTED SELECTION: A FAST TRACK TO INCREASE GENETIC GAIN IN PLANT AND ANIMAL BREEDING? SESSION II: MAS IN ANIMALS ANIMAL BREEDING FROM INFINITESIMAL MODEL TO MAS: THE CASE OF A BACKCROSS DESIGN IN DAIRY SHEEP (SARDA X LACAUNE) AND ITS POSSIBLE IMPACT ON SELECTION G. Pagnacco and A. Carta [email protected] [email protected] Summary Despite the great advancements achieved by quantitative techniques, selection in some species and for some traits, as in dairy sheep, can be very expensive and requires great efforts. In this paper we review the options offered by quantitative methods and by marker-assisted selection and we discuss the possible impact of a QTL detection design based on a backcross experimental population (Sarda X Lacaune) on sheep breeding. Keywords Infinitesimal model, Quantitative Trait Loci, Marker-assisted selection, dairy sheep, Backcross Design Abstract Selection under the infinitesimal model Standard animal breeding techniques have been largely successful in improving the performance of domestic animals in the last century. The success of these techniques finds its roots in the theoretical work of Fisher and Wright who showed how selection, migration and mutation could be derived from the simple Mendelian laws. All that theory was then transferred into the operational application in animal breeding by J. L. Lush (Animal Breeding Plans, 1945) and by his many “sons” and “grandsons”. In more recent times another landmark book, Introduction to Quantitative Genetics, (Falconer, 1960) probably represents the official birth of the quantitative genetics in plant and especially animal breeding. Plant and animal breeding have based their tools of investigation on this “central paradigm for the analysis of phenotypic variation” (Lynch and Walsh, 1998) even if their methods have radically diverged probably because of the different population structure in crop and domestic animals. -

Supplementary Information for Vyssotski, 2013
Supplementary Information for Vyssotski, 2013 www.evolocus.com/evolocus/v1/evolocus-01-013-s.pdf Evolocus Supplementary Information for Transgenerational epigenetic compensation and sex u al d imorph ism 1,2,3 Dmitri L. Vyssotski Published 17 May 2013, Evolocus 1, 13-18 (2013): http://www.evolocus.com/evolocus/v1/evolocus-01-013.pdf In natu ral popu lation transgenerational epigenetic compensation is by unknown mechanisms, the mechanisms those were difficult to generated in h omoz y gou s mu tants, mainly in males. It d oes not imagine, and this possibility was abandoned for over 47 years. immed iately affect th e ph enoty pe of h omoz y gou s mu tants, w h ere The higher variability in males can be a result of a not so good it w as generated . Transgenerational epigenetic compensation is canalization of their ontogenesis, and the canalization of localiz ed not in th e same locu s as original mu tation and it is ontogenesis is modulated by sex hormones. Thus, we can d ominant, b ecau se it affects th e ph enoty pe ev en if it is receiv ed imagine that the canalization of ontogenesis in males is from one parent. In fu rth er generations, starting from F 2, it is more artificially decreased in comparison with females by means of ex pressed in females th an in males: it increases fitness of hormonal differences. h omozy gou s mu tant females, d ecreases fitness of w ild ty pe The shift of the female distribution to the right was not females and h as w eak effect on h eterozy gou s females.