A Taxonomy and Genome Cartology of Maize Ltr Retrotransposons
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
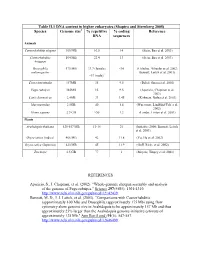
Table II.1 DNA Content in Higher Eukaryotes (Shapiro and Sternberg 2005) Species Genome Size % Repetitive DNA % Coding Sequences
Table II.1 DNA content in higher eukaryotes (Shapiro and Sternberg 2005) Species Genome size1 % repetitive % coding Reference DNA sequences Animals Caenorhabditis elegans 100 MB 16.5 14 (Stein, Bao et al. 2003) Caenorhabditis 104 MB 22.4 13 (Stein, Bao et al. 2003) briggsae Drosophila 175 MB 33.7 (female) <10 (Celniker, Wheeler et al. 2002; melanogaster Bennett, Leitch et al. 2003) ~57 (male)2 Ciona intestinalis 157MB 35 9.5 (Dehal, Satou et al. 2002) Fugu rubripes 365MB 15 9.5 (Aparicio, Chapman et al. 2002) Canis domesticus 2.4GB 31 1.45 (Kirkness, Bafna et al. 2003) Mus musculus 2.5GB 40 1.4 (Waterston, Lindblad-Toh et al. 2002) Homo sapiens 2.9 GB >50 1.2 (Lander, Linton et al. 2001) Plants Arabidopsis thaliana 125-157 MB 13-14 21 (Initiative 2000; Bennett, Leitch et al. 2003) Oryza sativa (indica) 466 MB 42 11.8 (Yu, Hu et al. 2002) Oryza sativa (Japonica) 420 MB 45 11.9 (Goff, Ricke et al. 2002) Zea mays 2.5 GB 77 1 (Meyers, Tingey et al. 2001) REFERENCES Aparicio, S., J. Chapman, et al. (2002). "Whole-genome shotgun assembly and analysis of the genome of Fugu rubripes." Science 297(5585): 1301-1310. http://www.ncbi.nlm.nih.gov/pubmed/12142439. Bennett, M. D., I. J. Leitch, et al. (2003). "Comparisons with Caenorhabditis (approximately 100 Mb) and Drosophila (approximately 175 Mb) using flow cytometry show genome size in Arabidopsis to be approximately 157 Mb and thus approximately 25% larger than the Arabidopsis genome initiative estimate of approximately 125 Mb." Ann Bot (Lond) 91(5): 547-557. -

The Repetitive Landscape of the 5100 Mbp Barley Genome Thomas Wicker1*, Alan H
Wicker et al. Mobile DNA (2017) 8:22 DOI 10.1186/s13100-017-0102-3 RESEARCH Open Access The repetitive landscape of the 5100 Mbp barley genome Thomas Wicker1*, Alan H. Schulman2,3, Jaakko Tanskanen2,3, Manuel Spannagl4, Sven Twardziok4, Martin Mascher5,6, Nathan M. Springer7, Qing Li7,8, Robbie Waugh9,10, Chengdao Li11,12, Guoping Zhang13, Nils Stein5, Klaus F. X. Mayer4,14 and Heidrun Gundlach4 Abstract Background: While transposable elements (TEs) comprise the bulk of plant genomic DNA, how they contribute to genome structure and organization is still poorly understood. Especially in large genomes where TEs make the majority of genomic DNA, it is still unclear whether TEs target specific chromosomal regions or whether they simply accumulate where they are best tolerated. Results: Here, we present an analysis of the repetitive fraction of the 5100 Mb barley genome, the largest angiosperm genome to have a near-complete sequence assembly. Genes make only about 2% of the genome, while over 80% is derived from TEs. The TE fraction is composed of at least 350 different families. However, 50% of the genome is comprised of only 15 high-copy TE families, while all other TE families are present in moderate or low copy numbers. We found that the barley genome is highly compartmentalized with different types of TEs occupying different chromosomal “niches”, such as distal, interstitial, or proximal regions of chromosome arms. Furthermore, gene space represents its own distinct genomic compartment that is enriched in small non-autonomous DNA transposons, suggesting that these TEs specifically target promoters and downstream regions. -

Factors That Affect Targeting of Ty5 Retrotransposon in Saccharomyces Cerevisiae Robert Dick Iowa State University
Iowa State University Capstones, Theses and Retrospective Theses and Dissertations Dissertations 2007 Factors that affect targeting of Ty5 retrotransposon in Saccharomyces cerevisiae Robert Dick Iowa State University Follow this and additional works at: https://lib.dr.iastate.edu/rtd Part of the Genetics and Genomics Commons Recommended Citation Dick, Robert, "Factors that affect targeting of Ty5 retrotransposon in Saccharomyces cerevisiae" (2007). Retrospective Theses and Dissertations. 15047. https://lib.dr.iastate.edu/rtd/15047 This Thesis is brought to you for free and open access by the Iowa State University Capstones, Theses and Dissertations at Iowa State University Digital Repository. It has been accepted for inclusion in Retrospective Theses and Dissertations by an authorized administrator of Iowa State University Digital Repository. For more information, please contact [email protected]. Factors that affect targeting of Ty5 retrotransposon in Saccharomyces cerevisiae by Robert Dick A dissertation submitted to the graduate faculty in partial fulfillment of the requirements for the degree of MASTER OF SCIENCE Major: Interdepartmental Genetics Program of Study Committee: Daniel F. Voytas, Major Professor Linda Abrosio Diane Bassham Iowa State University Ames, Iowa 2007 Copyright Robert Dick, 2007. All rights reserved. UMI Number: 1446046 Copyright 2007 by Dick, Robert All rights reserved. ____________________________________________________________ UMI Microform 1446046 Copyright 2007 by ProQuest Information and Learning Company. -

The Mechanism of Integration Preference to Heterochromatin of Yeast Retrotransposon Ty5 Weiwu Xie Iowa State University
Iowa State University Capstones, Theses and Retrospective Theses and Dissertations Dissertations 2003 The mechanism of integration preference to heterochromatin of yeast retrotransposon Ty5 Weiwu Xie Iowa State University Follow this and additional works at: https://lib.dr.iastate.edu/rtd Part of the Genetics Commons, and the Molecular Biology Commons Recommended Citation Xie, Weiwu, "The mechanism of integration preference to heterochromatin of yeast retrotransposon Ty5 " (2003). Retrospective Theses and Dissertations. 1475. https://lib.dr.iastate.edu/rtd/1475 This Dissertation is brought to you for free and open access by the Iowa State University Capstones, Theses and Dissertations at Iowa State University Digital Repository. It has been accepted for inclusion in Retrospective Theses and Dissertations by an authorized administrator of Iowa State University Digital Repository. For more information, please contact [email protected]. The mechanism of integration preference to heterochromatin of yeast retrotransposon Ty5 by Weiwu Xie A dissertation submitted to the graduate faculty in partial fulfillment of the requirements for the degree of DOCTOR OF PHILOSOPHY Major: Genetics (Computational Molecular Biology) Program of Study Committee: Daniel F. Voytas, Major Professor John May field Alan Myers David Oliver Jo Anne Powell-Coffman Iowa State University Ames, Iowa 2003 UMI Number: 3105118 UMI UMI Microform 3105118 Copyright 2003 by ProQuest Information and Learning Company. All rights reserved. This microform edition is protected against unauthorized copying under Title 17, United States Code. ProQuest Information and Learning Company 300 North Zeeb Road P.O. Box 1346 Ann Arbor, Ml 48106-1346 ii Graduate College Iowa State University This is to certify that the doctoral dissertation of Weiwu Xie Has met the dissertation requirements of Iowa State University Signature was redacted for privacy. -

Integration Specificity of the Retrovirus-Like Transposable Element Ty5 of Saccharomyces Sige Zou Iowa State University
Iowa State University Capstones, Theses and Retrospective Theses and Dissertations Dissertations 1996 Integration specificity of the retrovirus-like transposable element Ty5 of Saccharomyces Sige Zou Iowa State University Follow this and additional works at: https://lib.dr.iastate.edu/rtd Part of the Genetics Commons, Microbiology Commons, and the Molecular Biology Commons Recommended Citation Zou, Sige, "Integration specificity of the retrovirus-like transposable element Ty5 of Saccharomyces " (1996). Retrospective Theses and Dissertations. 11353. https://lib.dr.iastate.edu/rtd/11353 This Dissertation is brought to you for free and open access by the Iowa State University Capstones, Theses and Dissertations at Iowa State University Digital Repository. It has been accepted for inclusion in Retrospective Theses and Dissertations by an authorized administrator of Iowa State University Digital Repository. For more information, please contact [email protected]. INFORMATION TO USERS This manuscript has been reproduced from the microfilm master. UMI fihns the text directly fiom the original or copy submitted. Thus, some theas and dissertation copies are in typewriter &ce, vtliile others may be from ai^ type of computer printer. The quality of this reproduction is dependent upon the quality of the copy submitted. Broken or indistinct print, colored or poor quality illustrations and photographs, print bleedthrough, substandard margins, and improper alignment can adversely affect rq)roduction. In the unlikdy event that the author did not send UMI a complete nuuiuscript and there are misdng pages, these will be noted. Also, if unauthorized copyright material had to be removed, a note will indicate the deletion. Oversize materials (e.g., m^s, drawings, charts) are reproduced by sectioning the original, beginning at the upper left-hand comer and continuing from left to right in equal sections with small overlaps. -

Access to DNA Establishes a Secondary Target Site Bias for The
FEATURE Access to DNA establishes a secondary target site bias SACKLER SPECIAL for the yeast retrotransposon Ty5 Joshua A. Baller1, Jiquan Gao1, and Daniel F. Voytas2 Department of Genetics, Cell Biology, and Development, and Center for Genome Engineering, University of Minnesota, Minneapolis, MN 55455 Edited by Joan Curcio, Wadsworth Center, New York State Department of Health, Albany, NY, and accepted by the Editorial Board June 28, 2011 (received for review March 11, 2011) Integration sites for many retrotransposons and retroviruses are and HMR) (13–15). Ty5 IN selects integration sites using a 6-aa determined by interactions between retroelement-encoded inte- motif at the IN C terminus (16, 17). This IN targeting domain grases and specific DNA-bound proteins. The Saccharomyces interacts with a protein component of heterochromatin, namely retrotransposon Ty5 preferentially integrates into heterochroma- silent information regulator 4 (Sir4) (16, 18). The Ty5 IN/Sir4 tin because of interactions between Ty5 integrase and the interaction tethers the integration complex to target sites and heterochromatin protein silent information regulator 4. We map- results in the primary target site bias of Ty5. ped over 14,000 Ty5 insertions onto the S. cerevisiae genome, 76% In this study, we applied high-throughput DNA sequencing to of which occurred in heterochromatin, which is consistent with the characterize a large number of Ty5 insertions that we mapped to known target site bias of Ty5. Using logistic regression, associa- the S. cerevisiae genome. Whereas the majority of Ty5 elements tions were assessed between Ty5 insertions and various chromo- integrated as predicted in heterochromatin, a secondary target somal features such as genome-wide distributions of nucleosomes site bias was revealed for both euchromatic and heterochromatic and histone modifications. -

Retrovirus Interface: Gypsy and Copia Families in Yeast and Drosophila S.B
Morphogenesis at the Retrotransposon Retrovirus Interface: Gypsy and Copia Families in Yeast and Drosophila S.B. SANDMEYER and T.M. MENEES 1 Introduction . • . • . 261 2 Properties of Particles . • . 263 2.1 Copia Particles . • . 266 2.2 Copialike Ty Particles . • . 267 2.3 Drosophila Gypsy and Gypsylike Elements . • . • 272 2.4 Gypsylike Ty Particles . • . 274 3 Control of Relative Amounts of Structural and Catalytic Proteins . • 276 3.1 The -1 Frameshift Elements . • . 277 3.2 Plus 1 Frameshifting of Tyl . 277 3.3 Plus 1 Frameshift by the Incoming tRNA""' in Ty3 . • 278 3.4 Regulated Translation of Major Structural Proteins from L TR Retrotransposons with Single ORFs . • . 279 4 Priming and Reverse Transcription of LTR Retrotransposons . 280 4.1 Plus and M inus Strand Primers for Reverse Transcription . 280 4.2 Gypsy Gypsylike and Ty Reverse Transcription Intermediates . • . • . 282 4.3 Ty Priming by Initiator tRNAM•t . 283 4.4 Genetic Evidence that the tRNA Stem Is Not Copied into the Productive Plus-Strand Strong-Stop Species . • . • 284 4.5 Tyl Replication Intermediates . • . • . • . • . 285 4.6 Regulation of Ty3 Replication . • . • . • . • . 286 5 Regulation of Turnover of Ty Particles . • . 287 5.1 Turnover of Tyl Particles in Pheromone Arrested Cells . • . 287 5.2 Turnover of TY3 Particles by Components of the Stress Response . • 288 6 Application of Particles Based on Heterologous Ty Fusions 289 7 Prospects .. .. .... ... ... .. .. .... .. ...... • . • • • • • • • • • • • • • • • · · · · • 290 References . .. .•. ... ... ..•... •... , . • . • . 291 1 Introduction Long terminal repeat (LTR) retrotransposons have been identified in a wide variety of organisms including fungi (BoEKE and SANDMEYER 1991), plants (VoYTAS and AusueEL 1988; FLAVELL et al. 1995; SMYTH et al. -

Campus Veterinaire De Lyon
VETAGRO SUP CAMPUS VETERINAIRE DE LYON Année 2018 - Thèse n°089 ETUDE DE L'EVOLUTION D'UN ELEMENT TRANSPOSABLE DU GENOME DE LA CHAUVE-SOURIS PTEROPUS GIGANTEUS, ESPECE RESERVOIR DE ZOONOSES VIRALES THESE Présentée à l’UNIVERSITE CLAUDE-BERNARD - LYON I (Médecine - Pharmacie) et soutenue publiquement le 23 novembre 2018 pour obtenir le grade de Docteur Vétérinaire par COLLET Mathilde Née le 01 août 1993 à Voiron (38) VETAGRO SUP CAMPUS VETERINAIRE DE LYON Année 2018 - Thèse n°089 ETUDE DE L'EVOLUTION D'UN ELEMENT TRANSPOSABLE DU GENOME DE LA CHAUVE-SOURIS PTEROPUS GIGANTEUS, ESPECE RESERVOIR DE ZOONOSES VIRALES THESE Présentée à l’UNIVERSITE CLAUDE-BERNARD - LYON I (Médecine - Pharmacie) et soutenue publiquement le 23 novembre 2018 pour obtenir le grade de Docteur Vétérinaire par COLLET Mathilde Née le 1er août 1993 à Voiron (38) 1 2 Liste des Enseignants du Campus Vétérinaire de Lyon (1er mars 2018) Nom Prénom Département Grade ABITBOL Marie DEPT-BASIC-SCIENCES Maître de conférences ALVES-DE-OLIVEIRA Laurent DEPT-BASIC-SCIENCES Maître de conférences ARCANGIOLI Marie-Anne DEPT-ELEVAGE-SPV Professeur AYRAL Florence DEPT-ELEVAGE-SPV Maître de conférences BECKER Claire DEPT-ELEVAGE-SPV Maître de conférences BELLUCO Sara DEPT-AC-LOISIR-SPORT Maître de conférences BENAMOU-SMITH Agnès DEPT-AC-LOISIR-SPORT Maître de conférences BENOIT Etienne DEPT-BASIC-SCIENCES Professeur BERNY Philippe DEPT-BASIC-SCIENCES Professeur BONNET-GARIN Jeanne-Marie DEPT-BASIC-SCIENCES Professeur BOULOCHER Caroline DEPT-BASIC-SCIENCES Maître de conférences BOURDOISEAU -

The Saccharomyces Retrotransposon Ty5 Integrates Preferentially Into Regions of Silent Cfiromatin at the Telomeres and Mating Loci
Downloaded from genesdev.cshlp.org on September 28, 2021 - Published by Cold Spring Harbor Laboratory Press The Saccharomyces retrotransposon Ty5 integrates preferentially into regions of silent cfiromatin at the telomeres and mating loci Sige Zou, Ning Ke, Jin M. Kim, and Daniel F. Voytas 1 Department of Zoology and Genetics, Iowa State University, Ames, Iowa 50011 USA The nonrandom integration of retrotransposons and retroviruses suggests that chromatin influences target choice. Targeted integration, in turn, likely affects genome organization. In Saccharomyces, native Ty5 retrotransposons are located near telomeres and the silent mating locus HMR. To determine whether this distribution is a consequence of targeted integration, we isolated a transposition-competent Ty5 element from S. paradoxus, a species closely related to S. cerevisiae. This Ty5 element was used to develop a transposition assay in S. cerevisiae to investigate target preference of de novo transposition events. Of 87 independent Ty5 insertions, -30% were located on chromosome III, indicating this small chromosome (-1/40 of the yeast genome) is a highly preferred target. Mapping of the exact location of 19 chromosome III insertions showed that 18 were within or adjacent to transcriptional silencers flanking HML and HMR or the type X subtelomeric repeat. We predict Ty5 target preference is attributable to interactions between transposition intermediates and constituents of silent chromatin assembled at these sites. Ty5 target preference extends the link between telomere structure and reverse transcription as carried out by telomerase and Drosophila retrotransposons. [Key Words: Retrotransposon; Ty5; integration; transposition; silent chromatin; telomere] Received November 17, 1995; revised version accepted January 19, 1996. Mobile genetic elements that replicate by reverse tran- myces cerevisiae. -

Access to DNA Establishes a Secondary Target Site Bias for The
FEATURE Access to DNA establishes a secondary target site bias SACKLER SPECIAL for the yeast retrotransposon Ty5 Joshua A. Baller1, Jiquan Gao1, and Daniel F. Voytas2 Department of Genetics, Cell Biology, and Development, and Center for Genome Engineering, University of Minnesota, Minneapolis, MN 55455 Edited by Joan Curcio, Wadsworth Center, New York State Department of Health, Albany, NY, and accepted by the Editorial Board June 28, 2011 (received for review March 11, 2011) Integration sites for many retrotransposons and retroviruses are and HMR) (13–15). Ty5 IN selects integration sites using a 6-aa determined by interactions between retroelement-encoded inte- motif at the IN C terminus (16, 17). This IN targeting domain grases and specific DNA-bound proteins. The Saccharomyces interacts with a protein component of heterochromatin, namely retrotransposon Ty5 preferentially integrates into heterochroma- silent information regulator 4 (Sir4) (16, 18). The Ty5 IN/Sir4 tin because of interactions between Ty5 integrase and the interaction tethers the integration complex to target sites and heterochromatin protein silent information regulator 4. We map- results in the primary target site bias of Ty5. ped over 14,000 Ty5 insertions onto the S. cerevisiae genome, 76% In this study, we applied high-throughput DNA sequencing to of which occurred in heterochromatin, which is consistent with the characterize a large number of Ty5 insertions that we mapped to known target site bias of Ty5. Using logistic regression, associa- the S. cerevisiae genome. Whereas the majority of Ty5 elements tions were assessed between Ty5 insertions and various chromo- integrated as predicted in heterochromatin, a secondary target somal features such as genome-wide distributions of nucleosomes site bias was revealed for both euchromatic and heterochromatic and histone modifications. -

Controlled Integration of the Ty5 Retrotransposon in Saccharomyces Ceverisiae [I.E
Iowa State University Capstones, Theses and Retrospective Theses and Dissertations Dissertations 2006 Controlled integration of the Ty5 retrotransposon in Saccharomyces ceverisiae [i.e. cerevisiae] Junbiao Dai Iowa State University Follow this and additional works at: https://lib.dr.iastate.edu/rtd Part of the Genetics Commons Recommended Citation Dai, Junbiao, "Controlled integration of the Ty5 retrotransposon in Saccharomyces ceverisiae [i.e. cerevisiae] " (2006). Retrospective Theses and Dissertations. 1502. https://lib.dr.iastate.edu/rtd/1502 This Dissertation is brought to you for free and open access by the Iowa State University Capstones, Theses and Dissertations at Iowa State University Digital Repository. It has been accepted for inclusion in Retrospective Theses and Dissertations by an authorized administrator of Iowa State University Digital Repository. For more information, please contact [email protected]. Controlled integration of the Ty5 retrotransposon in Saccharomyces by Junbiao Dai A dissertation submitted to the graduate faculty in partial fulfillment of the requirements for the degree of DOCTOR OF PHILOSOPHY Major: Molecular, Cellular and Developmental Biology Program of Study Committee: Daniel F. Voytas, Major Professor Linda Ambrosio Janice Buss Michael Shogren-Knaak Yanhai Yin Iowa State University Ames, Iowa 2006 Copyright © Junbiao Dai, 2006. All rights reserved. UMI Number: 3229063 INFORMATION TO USERS The quality of this reproduction is dependent upon the quality of the copy submitted. Broken or indistinct print, colored or poor quality illustrations and photographs, print bleed-through, substandard margins, and improper alignment can adversely affect reproduction. In the unlikely event that the author did not send a complete manuscript and there are missing pages, these will be noted. -

Modeling Distributions of Chromosomal Modifications Using Chromosomal Features
Modeling Distributions of Chromosomal Modifications Using Chromosomal Features A DISSERTATION SUBMITTED TO THE FACULTY OF THE GRADUATE SCHOOL OF THE UNIVERSITY OF MINNESOTA BY Joshua Adam Baller IN PARTIAL FULFILLMENT OF THE REQUIREMENTS FOR THE DEGREE OF DOCTOR OF PHILOSOPHY Chad Myers, Daniel Voytas February 2012 Copyright Joshua A. Baller 2012 Acknowledgements J. Gao played an integral role in the production of the Ty5 and Ty1 datasets. H. Wang, D. Mayhew and R. Mitra made additional Ty5 data available prior to publication. M. Hickman made K. lactis ORC data available prior to publication Also, thanks to J. Carlis for advice on data storage as well as to R. Bushman and N. Milani for advice on data processing and statistical approaches. i This dissertation is dedicated to: My mother, father and sister You have given me a great deal of love and support. This dissertation is the culmination of years of work, long nights and little free time. I wouldn’t have made it to this point without your patience and understanding. My thesis advisors: Dan, Chad and (honorarily) Judy These discoveries were not made in a vacuum. You have been my sounding boards, my strongest allies and my harshest critics. I hope in my own career I can be equal to your examples. And to all my mentors and advisors throughout the years Your encouragement and advice inspired me to push my limits and discover new frontiers. ii Abstract Chromatin plays a major role in the regulation and evolution of genomic DNA. The advent of high-throughput sequencing, and the subsequently increasing availability of sequencing data from chromatin immunoprecipitation experiments, is leading to a comprehensive view of the chromatin landscape in key model organisms such as S.