Hypomethylation-Linked Activation of PLCE1 Impedes Autophagy and Promotes Tumorigenesis Through MDM2-Mediated Ubiquitination and Destabilization of P53
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Deciphering the Functions of Ets2, Pten and P53 in Stromal Fibroblasts in Multiple
Deciphering the Functions of Ets2, Pten and p53 in Stromal Fibroblasts in Multiple Breast Cancer Models DISSERTATION Presented in Partial Fulfillment of the Requirements for the Degree Doctor of Philosophy in the Graduate School of The Ohio State University By Julie Wallace Graduate Program in Molecular, Cellular and Developmental Biology The Ohio State University 2013 Dissertation Committee: Michael C. Ostrowski, PhD, Advisor Gustavo Leone, PhD Denis Guttridge, PhD Dawn Chandler, PhD Copyright by Julie Wallace 2013 Abstract Breast cancer is the second most common cancer in American women, and is also the second leading cause of cancer death in women. It is estimated that nearly a quarter of a million new cases of invasive breast cancer will be diagnosed in women in the United States this year, and approximately 40,000 of these women will die from breast cancer. Although death rates have been on the decline for the past decade, there is still much we need to learn about this disease to improve prevention, detection and treatment strategies. The majority of early studies have focused on the malignant tumor cells themselves, and much has been learned concerning mutations, amplifications and other genetic and epigenetic alterations of these cells. However more recent work has acknowledged the strong influence of tumor stroma on the initiation, progression and recurrence of cancer. Under normal conditions this stroma has been shown to have protective effects against tumorigenesis, however the transformation of tumor cells manipulates this surrounding environment to actually promote malignancy. Fibroblasts in particular make up a significant portion of this stroma, and have been shown to impact various aspects of tumor cell biology. -

A Cytoskeleton Regulator AVIL Drives Tumorigenesis in Glioblastoma
ARTICLE https://doi.org/10.1038/s41467-020-17279-1 OPEN A cytoskeleton regulator AVIL drives tumorigenesis in glioblastoma Zhongqiu Xie 1, Pawel Ł. Janczyk2,8, Ying Zhang3,8, Aiqun Liu4, Xinrui Shi1, Sandeep Singh 1, Loryn Facemire1, ✉ Kristopher Kubow5,ZiLi6, Yuemeng Jia1, Dorothy Schafer7, James W. Mandell1, Roger Abounader3 & Hui Li1,2 Glioblastoma is a deadly cancer, with no effective therapies. Better understanding and identification of selective targets are urgently needed. We found that advillin (AVIL) is 1234567890():,; overexpressed in all the glioblastomas we tested including glioblastoma stem/initiating cells, but hardly detectable in non-neoplastic astrocytes, neural stem cells or normal brain. Glioma patients with increased AVIL expression have a worse prognosis. Silencing AVIL nearly eradicated glioblastoma cells in culture, and dramatically inhibited in vivo xenografts in mice, but had no effect on normal control cells. Conversely, overexpressing AVIL promoted cell proliferation and migration, enabled fibroblasts to escape contact inhibition, and transformed immortalized astrocytes, supporting AVIL being a bona fide oncogene. We provide evidence that the tumorigenic effect of AVIL is partly mediated by FOXM1, which regulates LIN28B, whose expression also correlates with clinical prognosis. AVIL regulates the cytoskeleton through modulating F-actin, while mutants disrupting F-actin binding are defective in its tumorigenic capabilities. 1 Department of Pathology, School of Medicine, University of Virginia, Charlottesville, VA 22908, USA. 2 Department of Biochemistry and Molecular Genetics, School of Medicine, University of Virginia, Charlottesville, VA 22908, USA. 3 Department of Microbiology, Immunology, and Cancer Biology, School of Medicine, University of Virginia, Charlottesville, VA 22908, USA. 4 Tumor Hospital, Guangxi Medical University, Nanning 530021, China. -

Biogenesis and Maintenance of Cytoplasmic Domains in Myelin of the Central Nervous System
Biogenesis and Maintenance of Cytoplasmic Domains in Myelin of the Central Nervous System Dissertation for the award of the degree “Doctor rerum naturalium” of the Georg-August-Universität Göttingen within the doctoral program Molecular Biology of Cells of the Georg-August University School of Science (GAUSS) submitted by Caroline Julia Velte from Usingen, Germany Göttingen 2016 Members of the Thesis Committee: Prof. Dr. Mikael Simons, Reviewer Max Planck Institute of Experimental Medicine, Göttingen Prof. Dr. Andreas Janshoff, Reviewer Georg-August University, Göttingen Prof. Dr. Dirk Görlich Max Planck Institute of Biophysical Chemistry, Göttingen Date of the thesis defense: 27th of June 2016 Don't part with your illusions. When they are gone, you may still exist, but you have ceased to live. (Mark Twain) III Affidavit I hereby declare that my PhD thesis “Biogenesis and Maintenance of Cytoplasmic Domains in Myelin of the Central Nervous System” has been written independently with no other aids or sources than quoted. Furthermore, I confirm that this thesis has not been submitted as part of another examination process neither in identical nor in similar form. Caroline Julia Velte April 2016 Göttingen, Germany V Publications Nicolas Snaidero*, Caroline Velte*, Matti Myllykoski, Arne Raasakka, Alexander Ignatev, Hauke B. Werner, Michelle S. Erwig, Wiebke Moebius, Petri Kursula, Klaus-Armin Nave, and Mikael Simons Antagonistic Functions of MBP and CNP Establish Cytosolic Channels in CNS Myelin, Cell Reports 18 *equal contribution (January 2017) E. d’Este, D. Kamin, C. Velte, F. Göttfert, M. Simons, S.W. Hell Subcortical cytoskeleton periodicity throughout the nervous system, Scientific Reports 6 (March 2016) K.A. -

Phospholipase C-Related Catalytically Inactive Protein: a Novel Signaling Molecule for Modulating Fat Metabolism and Energy Expenditure
Journal of Oral Biosciences 61 (2019) 65e72 Contents lists available at ScienceDirect Journal of Oral Biosciences journal homepage: www.elsevier.com/locate/job Review Phospholipase C-related catalytically inactive protein: A novel signaling molecule for modulating fat metabolism and energy expenditure * Takashi Kanematsu a, b, , Kana Oue a, c, Toshiya Okumura a, Kae Harada a, 1, Yosuke Yamawaki a, 2, Satoshi Asano a, Akiko Mizokami d, Masahiro Irifune c, Masato Hirata e a Department of Cellular and Molecular Pharmacology, Division of Basic Life Sciences, Institute of Biomedical and Health Sciences, Hiroshima University, Hiroshima, 734-8553, Japan b Department of Cell Biology and Pharmacology, Faculty of Dental Science, Kyushu University, Fukuoka, 812-8582, Japan c Department of Dental Anesthesiology, Division of Applied Life Sciences, Institute of Biomedical and Health Sciences, Hiroshima University, Hiroshima, 734- 8553, Japan d OBT Research Center, Faculty of Dental Science, Kyushu University, Fukuoka, 812-8582, Japan e Fukuoka Dental College, Fukuoka, 814-0193, Japan article info abstract Article history: Background: Overweight and obesity are defined as excessive or abnormal fat accumulation in adipose Received 16 March 2019 tissues, and increase the risk of morbidity in many diseases, including hypertension, dyslipidemia, type 2 Received in revised form diabetes, coronary heart disease, and stroke, through pathophysiological mechanisms. There is strong 17 April 2019 evidence that weight loss reduces the risk of metabolic syndrome by limiting blood pressure and Accepted 19 April 2019 improving the levels of serum triglycerides, total cholesterol, low-density lipoprotein-cholesterol, and Available online 15 May 2019 high-density lipoprotein-cholesterol. To date, several attempts have been made to develop effective anti- obesity medication or weight-loss drugs; however, satisfactory drugs for clinical use have not yet been Keywords: Adipose tissue developed. -

Exome Sequencing Reveals Cubilin Mutation As a Single-Gene Cause of Proteinuria
BRIEF COMMUNICATION www.jasn.org Exome Sequencing Reveals Cubilin Mutation as a Single-Gene Cause of Proteinuria Bugsu Ovunc,*† Edgar A. Otto,* Virginia Vega-Warner,* Pawaree Saisawat,* Shazia Ashraf,* Gokul Ramaswami,* Hanan M. Fathy,‡ Dominik Schoeb,* Gil Chernin,* Robert H. Lyons,§ ʈ Engin Yilmaz,† and Friedhelm Hildebrandt* ¶ ʈ Departments of *Pediatrics and Human Genetics, §Department of Biological Chemistry and DNA Sequencing Core, and ¶Howard Hughes Medical Institute, University of Michigan, Ann Arbor, Michigan; †Department of Medical Biology, Hacettepe University, Ankara, Turkey; and ‡The Pediatric Nephrology Unit, Alexandria University, Alexandria, Egypt ABSTRACT In two siblings of consanguineous parents with intermittent nephrotic-range pro- tion is still unknown.7 This forbids the use of teinuria, we identified a homozygous deleterious frameshift mutation in the gene cohort studies for gene identification and ne- CUBN, which encodes cubulin, using exome capture and massively parallel re- cessitates the ability to identify disease-caus- sequencing. The mutation segregated with affected members of this family and ing genes in single families. We therefore was absent from 92 healthy individuals, thereby identifying a recessive mutation in combined whole genome homozygosity CUBN as the single-gene cause of proteinuria in this sibship. Cubulin mutations mapping with consecutive whole human ex- cause a hereditary form of megaloblastic anemia secondary to vitamin B12 defi- ome capture (WHEC) and massively par- ciency, and proteinuria occurs in 50% of cases since cubilin is coreceptor for both allel re-sequencing to overcome this lim- 6 the intestinal vitamin B12-intrinsic factor complex and the tubular reabsorption of itation. In this way we here identify a protein in the proximal tubule. -

140503 IPF Signatures Supplement Withfigs Thorax
Supplementary material for Heterogeneous gene expression signatures correspond to distinct lung pathologies and biomarkers of disease severity in idiopathic pulmonary fibrosis Daryle J. DePianto1*, Sanjay Chandriani1⌘*, Alexander R. Abbas1, Guiquan Jia1, Elsa N. N’Diaye1, Patrick Caplazi1, Steven E. Kauder1, Sabyasachi Biswas1, Satyajit K. Karnik1#, Connie Ha1, Zora Modrusan1, Michael A. Matthay2, Jasleen Kukreja3, Harold R. Collard2, Jackson G. Egen1, Paul J. Wolters2§, and Joseph R. Arron1§ 1Genentech Research and Early Development, South San Francisco, CA 2Department of Medicine, University of California, San Francisco, CA 3Department of Surgery, University of California, San Francisco, CA ⌘Current address: Novartis Institutes for Biomedical Research, Emeryville, CA. #Current address: Gilead Sciences, Foster City, CA. *DJD and SC contributed equally to this manuscript §PJW and JRA co-directed this project Address correspondence to Paul J. Wolters, MD University of California, San Francisco Department of Medicine Box 0111 San Francisco, CA 94143-0111 [email protected] or Joseph R. Arron, MD, PhD Genentech, Inc. MS 231C 1 DNA Way South San Francisco, CA 94080 [email protected] 1 METHODS Human lung tissue samples Tissues were obtained at UCSF from clinical samples from IPF patients at the time of biopsy or lung transplantation. All patients were seen at UCSF and the diagnosis of IPF was established through multidisciplinary review of clinical, radiological, and pathological data according to criteria established by the consensus classification of the American Thoracic Society (ATS) and European Respiratory Society (ERS), Japanese Respiratory Society (JRS), and the Latin American Thoracic Association (ALAT) (ref. 5 in main text). Non-diseased normal lung tissues were procured from lungs not used by the Northern California Transplant Donor Network. -

Stimulation of Phospholipid Metabolism in Embryonic Muscle
Proc. Natl. Acad. Sci. USA Vol. 76, No. 9, pp. 4474-4478, September 1979 Cell Biology Stimulation of phospholipid metabolism in embryonic muscle cells treated with phospholipase C (phospholipid synthesis/myogenesis) CLAUDIA KENT Department of Biochemistry, Purdue University, West Lafayette, Indiana 47907 Communicated by Edwin T. Mertz, May 29, 1979 ABSTRACT Phospholipid metabolism is dramatically MATERIALS AND METHODS stimulated in cultured myogenic cells in which cell fusion was inhibited with phospholipase C (phosphatidylcholine choline- Cultured Cells. Pectoral muscle from 11-day chicken em- phosphohydrolase; EC 3.1.4.3). Phospholipase C was active bryos was dissected, loose connective tissue was removed, and under the culture conditions as shown by the degradation of the muscle was minced into 1- to 2-mm fragments. Cells were exogenous phosphatidylcholine. Rates of incorporation of 32p; dissociated from the tissue fragments by trituration with a and [metkyl-3Hlcholine into lipids were about 5-fold greater in phospholipase-treated cells than in either untreated fusing cells pasteur pipette (8) in calcium- and magnesium-free Earle's salt or untreated cells prevented from fusing by-calcium deprivation. solution. The cell suspension was filtered through cheesecloth, The greatest stimulation in the phospholipase C-treated cultures preplated for 15 min (9), and then diluted with culture medium occurred with synthesis of phospai tlcholine and sphin- to 5 X 105 cells/ml. The cells were plated in tissue culture dishes gomyelin; synthesis of phosphatidyinositol and cardiolipin was precoated with rat tail collagen (10) at 8 ml of cell suspension not stimulated. Degradation of cellular [32Plphosphatidylcholine and appearance in the culture medium of the degradation per 100 mm dish. -

Disruption of Epithelial Architecture Caused by Loss of PTEN Or by Oncogenic Mutant P110a/PIK3CA but Not by HER2 Or Mutant AKT1
Oncogene (2013) 32, 4417–4426 & 2013 Macmillan Publishers Limited All rights reserved 0950-9232/13 www.nature.com/onc ORIGINAL ARTICLE Disruption of epithelial architecture caused by loss of PTEN or by oncogenic mutant p110a/PIK3CA but not by HER2 or mutant AKT1 FM Berglund1,3, NR Weerasinghe1,3, L Davidson1,3, JC Lim1,{, BJ Eickholt2 and NR Leslie1 Genetic changes in HER2, PTEN, PIK3CA and AKT1 are all common in breast cancer and lead to the elevated phosphorylation of downstream targets of the PI3K/AKT signalling pathway. Changes in HER2, PTEN, PIK3CA and AKT have all been reported to lead to both enhanced proliferation and failures in hollow lumen formation in three dimensional epithelial culture models, but it is not clear whether these failures in lumen formation are caused by any failure in the spatial coordination of lumen formation (hollowing) or purely a failure in the apoptosis and clearance of luminal cells (cavitation). Here, we use normal murine mammary gland (NMuMG) epithelial cells, which form a hollow lumen without significant apoptosis, to compare the transformation by these four genetic changes. We find that either mutant PIK3CA expression or PTEN loss, but not mutant AKT1 E17K, cause disrupted epithelial architecture, whereas HER2 overexpression drives strong proliferation without affecting lumen formation in these cells. We also show that PTEN requires both lipid and protein phosphatase activity, its extreme C-terminal PDZ binding sequence and probably Myosin 5A to control lumen formation through a mechanism that does not correlate with its ability to control AKT, but which is selectively lost through mutation in some tumours. -

A Role for Nuclear PTEN in Neuronal Differentiation
The Journal of Neuroscience, February 15, 2000, 20(4):1404–1413 A Role for Nuclear PTEN in Neuronal Differentiation Mahesh B. Lachyankar,1 Nazneen Sultana,1 Christopher M. Schonhoff,2 Prasenjit Mitra,1 Wojciech Poluha,1 Stephen Lambert,3 Peter J. Quesenberry,4 N. Scott Litofsky,5 Lawrence D. Recht,6 Roya Nabi,7 Susan J. Miller,8 Shinji Ohta,8 Benjamin G. Neel,8 and Alonzo H. Ross1 Departments of 1Pharmacology and Molecular Toxicology, 2Biochemistry and Molecular Biology, 3Cell Biology, 4Medicine (Cancer Center), 5Surgery, and 6Neurology, University of Massachusetts Medical School, Worcester, Massachusetts 01655, 7Department of Biology and Biotechnology, Worcester Polytechnic Institute, Worcester, Massachusetts 01609, and 8Cancer Biology Program, Division of Hematology-Oncology, Department of Medicine, Beth Israel-Deaconess Medical Center, Boston, Massachusetts 02215 Mutations of phosphatase and tensin homolog deleted on chro- detected in both the nucleus and cytoplasm. Suppression of mosome 10 (PTEN), a protein and lipid phosphatase, have been PTEN levels with antisense oligonucleotides does not block associated with gliomas, macrocephaly, and mental deficien- initiation of neuronal differentiation. Instead, PTEN antisense cies. We have assessed PTENЈs role in the nervous system and leads to death of the resulting, immature neurons, probably find that PTEN is expressed in mouse brain late in develop- during neurite extension. In contrast, PTEN is not required for ment, starting at approximately postnatal day 0. In adult brain, astrocytic differentiation. These observations indicate that PTEN is preferentially expressed in neurons and is especially PTEN acts at multiple sites in the cell, regulating the transition evident in Purkinje neurons, olfactory mitral neurons, and large of differentiating neuroblasts to postmitotic neurons. -

Truseq 48 Gene Cancer Panel
TruSeq 48 Gene Cancer Panel Indication: Next generation sequencing (NGS) assay for detection of hot spot mutations in 48 cancer-related genes: ABL1, AKT1, ALK, APC, ATM, BRAF, CDH1, CDKN2A, CSF1R, CTNNB1, EGFR, ERBB2, ERBB4, FBXW7, FGFR1, FGFR2, FGFR3, FLT3, GNA11, GNAQ, GNAS, HNF1A, HRAS, IDH1, JAK2, JAK3, KDR, KIT, KRAS, MET, MLH1, MPL, NOTCH1, NPM1, NRAS, PDGFRA, PIK3CA, PTEN, PTPN11, RB1, RET, SMAD4, SMARCB1, SMO, SRC, STK11, TP53 and VHL. The specific mutations are detected by amplification of the corresponding exons by polymerase chain reaction (PCR). The PCR product is sequenced on a Illumina's MiSeqDx NGS Sequencer. Annotation is provided by Cartagenia Bench Lab NGS. Clinical Panel Gene Exon / Amino Acid (AA) Coverage ABL1 Exons 4-7 (AA 243-362, 395-424) ALK Exons 23, 25 (AA 1172-1176, 1248-1275) BRAF Exon 15 (AA 581-606) EGFR Exons 3, 7, 18-21 (AA 108-142, 287-297, 598-627, 708-728, 729-761, 762-817, 857-875) GNA11 Exons 4-7 (AA 159-172, 172-202, 202-216, 255-297, 304-349, 349-360) GNAQ Exons 4- 7 (AA 159-202, 202-210, 210-263, 261-297, 297-325, 326-360, 355-360) IDH1 Exon 4 (AA 95-133) JAK2 Exon 14 (AA 615-622) KIT Exons 11, 13, 17 (AA 550-587, 641-664, 814-828) KRAS Exons 2-3 (AA 1-22, 38-63) MPL Exon 10 (AA 514-522) NRAS Exons 2-3 (AA 1-19, 38-62) NPM1 Exon 11 (AA 283-295) PIK3CA Exons 2, 5, 8, 10, 21 (AA 84-118, 345-353, 418-441, 538-555, 987-1068) Investigational Panel Gene Exon / Amino Acid (AA) Coverage AKT1 Exon 4 (AA 16-49) APC Exon 16 (AA 875-918, 1113-1153, 1257-1575) ATM Exons 8, 9, 12, 17, 26, 34-36, 50, 54-56, 59, -
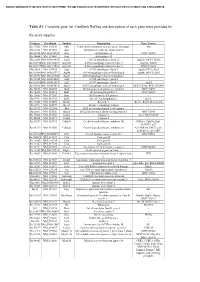
Table S1. Complete Gene List. Genbank Refseq and Description of Each Gene Were Provided By
Document downloaded from http://www.elsevier.es, day 24/09/2021. This copy is for personal use. Any transmission of this document by any media or format is strictly prohibited. Table S1. Complete gene list. GenBank RefSeq and description of each gene were provided by the array supplier. Unigene GeneBank Symbol Description Gene Name/s Rn.11422 NM_033230 Akt1 V-akt murine thymoma viral oncogene homolog 1 Akt Rn.2104 NM_019288 App Amyloid beta (A4) precursor protein - Rn.23323 NM_001034933 Arsa Arylsulfatase A MGC125207 Rn.94004 NM_033443 Arsb Arylsulfatase B - Rn.6224 NM_001038495 Atg12 ATG12 autophagy related 12 Apg12l, MGC125080 Rn.101734NM_001108809 Atg16l1 ATG16 autophagy related 16-like 1 Apg16l, Wdr30 Rn.104199NM_001191560 Atg16l2 ATG16 autophagy related 16-like 2 RGD1311400 Rn.3084 NM_134394 Atg3 ATG3 autophagy related 3 Apg3l, PIG-1, Pig1 Rn.163086NM_001025711 Atg4b ATG4 autophagy related 4 homolog B Apg4b, MGC112887 Rn.23378 NM_001107948 Atg4c ATG4 autophagy related 4 homolog C - Rn.98385 NM_001014250 Atg5 ATG5 autophagy related 5 - Rn.162765NM_001012097 Atg7 ATG7 autophagy related 7 Apg7l Rn.35248 NM_001014218 Atg9a ATG9 autophagy related 9 homolog A MGC105908, RGD1310450 Rn.36696 NM_022698 Bad BCL2-associated agonist of cell death MGC72439 Rn.14598 NM_053812 Bak1 BCL2-antagonist/killer 1 MGC108627 Rn.10668 NM_017059 Bax Bcl2-associated X protein - Rn.9996 NM_016993 Bcl2 B-cell CLL/lymphoma 2 Bcl-2 Rn.10323 NM_031535 Bcl2l1 Bcl2-like 1 Bcl-xl, Bcl2l, Bclx, bcl-X Rn.2776 NM_053739 Becn1 Beclin 1, autophagy related - Rn.31142 NM_022684 -

The Functions of Tumor Suppressor PTEN in Innate and Adaptive Immunity
Cellular & Molecular Immunology (2017) 14, 581–589 & 2017 CSI and USTC All rights reserved 2042-0226/17 $32.00 www.nature.com/cmi REVIEW The functions of tumor suppressor PTEN in innate and adaptive immunity Lang Chen1 and Deyin Guo2 The tumor suppressor phosphatase and tensin homolog (PTEN) is a lipid and protein phosphatase that is able to antagonize the PI3K/AKT pathway and inhibit tumor growth. PTEN also possesses phosphatase-independent functions. Genetic alterations of PTEN may lead to the deregulation of cell proliferation, survival, differentiation, energy metabolism and cellular architecture and mobility. Although the role of PTEN in tumor suppression is extensively documented and well established, the evidence for its roles in immunity did not start to accumulate until recently. In this review, we will focus on the newly discovered functions of PTEN in the regulation of innate and adaptive immunity, including antiviral responses. Cellular & Molecular Immunology (2017) 14, 581–589; doi:10.1038/cmi.2017.30; published online 26 June 2017 Keywords: adaptive immunity; innate immunity; PTEN; tumor suppressor PTEN AND TUMOR SUPPRESSION found in tumor cells (http://cancer.sanger.ac.uk/cosmic/search? In 1997, two groups independently found that there is a tumor q = pten). Some mutations can even directly change the suppressor gene located on chromosome 10q23.1,2 Li et al.1 conformation of PTEN or can impair its phosphatase activity. named the gene PTEN, abbreviated from phosphatase and Researchers have also discovered that mutation of one of the tensin homolog (PTEN) deleted on chromosome 10, which PTEN alleles could affect the function of PTEN.