Probing Molecular Dynamics with Non-Linear Optical Techniques
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Temporal and Spectral Characterization of Near-Ir Femtosecond Optical Parametric Oscillator Pulses
MSc in Photonics PHOTONICSBCN Universitat Politècnica de Catalunya (UPC) Universitat Autònoma de Barcelona (UAB) Universitat de Barcelona (UB) Institut de Ciències Fotòniques (ICFO) http://www.photonicsbcn.eu Master in Photonics MASTER THESIS WORK TEMPORAL AND SPECTRAL CHARACTERIZATION OF NEAR-IR FEMTOSECOND OPTICAL PARAMETRIC OSCILLATOR PULSES Parisa Farzam Supervised by Dr. Majid Ebrahim-Zadeh, (ICFO, ICREA) and Dr. Lukasz Kornaszewski (ICFO) Presented on date 9th September 2009 Registered at Temporal and spectral characterization of near-IR femtosecond optical parametric oscillator pulses Parisa Farzam ICFO-Institut de Ciències Fotòniques, Nonlinear optics group, Mediterranean Technology Park, 08860 Castelldefels (Barcelona), Spain E-mail: [email protected] Abstract. Recently, there has been growing interest in ultrashort pulse measurement techniques. The aim of this work is to utilize interferometric autocorrelator in order to characterize near- IR femtosecond optical parametric oscillator (OPO) pulses. Although considerable research has been devoted to using nonlinear crystals in the autocorrelators, an attractive alternative to this approach remains two-photon absorption in photodiodes. In the setup we have used a commercial semiconductor which has several advantages to second harmonic generation technique. Conventional methods in autocorrelation analysis use secant hyperbolic square (sech2) assumption to figure out the shape of pulse; but we have developed a software to find out the pulse shape by guessing the phase. The comparison between results from phase-guessing method and sech2 assumption shows that the first method is more flexible and gives much information but it needs to be enhanced in order to find the best phase automatically. Keywords: ultrashort pulse measurement, autocorrelation, two-photon absorption, optical parametric oscillator. -
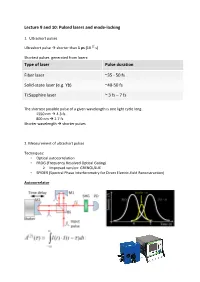
Lecture 9 and 10: Pulsed Lasers and Mode-Locking
Lecture 9 and 10: Pulsed lasers and mode-locking 1. Ultrashort pulses Ultrashort pulse shorter than 1 ps (10-12 s) Shortest pulses generated from lasers: Type of laser Pulse duration Fiber laser ~35 - 50 fs Solid-state laser (e.g. Yb) ~40-50 fs Ti:Sapphire laser ~ 3 fs – 7 fs The shortest possible pulse of a given wavelength is one light cycle long: 1550 nm 4.3 fs 800 nm 2.7 fs Shorter wavelength shorter pulses 2. Measurement of ultrashort pulses Techniques: • Optical autocorrelation • FROG (Frequency Resolved Optical Gating) Improved version: GRENOULLIE • SPIDER (Spectral Phase Interferometry for Direct Electric-field Reconstruction) Autocorrelator Interferometric autocorrelation trace: FROG Source: Rick Trebino, Frequency Resolved Optical Gating SPIDER 3. Mode-locked lasers a) Difference between a CW and mode-locked laser: A mode-locked laser: Emits a train of equally-spaced, ultrashort pulses (femtosecond) - down to almost single- cycle Output spectrum is broad (tens-hundreds of nanometers)/ Broader spectrum shorter pulses In the frequency domain, the output consists of thousands/millions of narrow lines (comb- like structure) b) Relation between the number of synchronized modes and the pulse duration: More synchronized modes shorter pulse. c) Passive mode-locking and saturable absorber In order to achieve passive mode-locking we need a device called saturable absorber. The saturable absorber: - Blocks any CW radiation in the cavity - It forces the laser to synchronize the modes and generate pulses - Only high-intensity pulse -

Nanoscale Force Sensing of an Ultrafast Nonlinear Optical Response
Nanoscale force sensing of an ultrafast nonlinear optical response Zeno Schumachera,1,2 , Rasa Rejalia, Raphael Pachlatkoa , Andreas Spielhofera, Philipp Naglerb, Yoichi Miyaharaa, David G. Cookea , and Peter Grutter¨ a,1 aDepartment of Physics, McGill University, Montreal QC H3A 2T8, Canada; and bDepartment of Physics, University of Regensburg, 93053 Regensburg, Germany Edited by Jianming Cao, Florida State University, Tallahassee, FL, and accepted by Editorial Board Member Zachary Fisk July 2, 2020 (received for review March 2, 2020) The nonlinear optical response of a material is a sensitive probe Results of electronic and structural dynamics under strong light fields. We split the output of a mode-locked femtosecond fiber laser The induced microscopic polarizations are usually detected via operating at 80-MHz repetition rate and 780-nm central wave- their far-field light emission, thus limiting spatial resolution. Sev- length, 200 mW (Toptica FemtoFiber pro NIR), to generate eral powerful near-field techniques circumvent this limitation two coherent pulse trains, with a well-defined temporal delay by employing local nanoscale scatterers; however, their signal between the two. The pulse trains can be arranged in a non- strength scales unfavorably as the probe volume decreases. Here, collinear or collinear fashion and focused onto the tip–sample we demonstrate that time-resolved atomic force microscopy is junction of an AFM incorporated in a UHV system. As the two capable of temporally and spatially resolving the microscopic, pulses are delayed with respect to each other, the light inten- electrostatic forces arising from a nonlinear optical polarization sity at the tip–sample junction oscillates, in delay time, with the in an insulating dielectric driven by femtosecond optical fields. -

Complete Characterization of Optical Pulses in the Picosecond Regime for Ultrafast Communication Systems by Jade P
-A Complete Characterization of Optical Pulses in the Picosecond Regime for Ultrafast Communication Systems by Jade P. Wang Submitted to the Department of Electrical Engineering and Computer Science in partial fulfillment of the requirements for the degree of Master of Engineering in Electrical Engineering and Computer Science at the MASSACHUSETTS INSTITUTE OF TECHNOLOGY August 2002 @ Massachusetts Institute of Technology, 2002. All rights reserved MASSACHUSETTS INSTITUTE OF TECHNOLOGY JUL 3 0 2003 LIBRARIES V, Author.. ..........................................- fDepart'rrisfit of Electrical Engineering and Computer Science August 21, 2002 Certified by........ ........................ Erich P. Ippen Elihu Thomson Professor of Electrical Engineering Thesis Supervisor Certified by....... ............................. Scott A. Hamilton MIT Lincoln Laboratory Staff Member T)p6s Supervisor Accepted by............. Arthur C. Smith Chairman, Deparment Committee on Graduate Theses 10411 2 Complete Characterization of Optical Pulses in the Picosecond Regime for Ultrafast Communication Systems by Jade P. Wang Submitted to the Department of Electrical Engineering and Computer Science in partial fulfillment of the requirements for the degree of Master of Engineering in Electrical Engineering and Computer Science Abstract Ultrashort optical pulses have a variety of applications, one of which is the develop- ment of optical time-devision multiplexing (OTDM) networks. Data is encoded in these short optical pulses (typically a few picoseconds in length) which are then inter- leaved in time to provide very high data rates on a single wavelength. Wavelength- division multiplexing (WDM) uses multiple channels by interleaving pulses in fre- quency in order to achieve high data rates. Typical pulse lengths in WDM are on the order of hundreds of picoseconds. OTDM networks have some advantages over WDM networks, but in order to take advantage of these characteristics, a better understanding of short optical pulse characterization needs to be reached. -

Towards On-Chip MEMS-Based Optical Autocorrelator
This is the author's version of an article that has been published in this journal. Changes were made to this version by the publisher prior to publication. The final version of record is available at http://dx.doi.org/10.1109/JLT.2018.2867473 > REPLACE THIS LINE WITH YOUR PAPER IDENTIFICATION NUMBER (DOUBLE-CLICK HERE TO EDIT) < 1 Towards On-Chip MEMS-Based Optical Autocorrelator Ahmed M. Othman, Student Member, IEEE, Hussein E. Kotb, Member, IEEE, Yasser M. Sabry, Member, IEEE, Osama Terra, and Diaa A. Khalil, Senior Member, IEEE have a very limited wavelength range of operation [7], or only Abstract—We propose a compact MEMS-based optical capable of measuring pulses having a time-bandwidth product autocorrelator based on a micromachined Michelson greater than 100 [8]. In addition, some work has been reported interferometer in silicon and the two-photon absorption non- based on CdS or CdTe nanowires [9]–[10] but the use of linearity in a photodetector. The miniaturized autocorrelator has alignment-sensitive optical components for coupling light into a scanning range of 1.2 ps and operates in the wavelength range of 1100-2000 nm. The device measures the interferometric the nanowires is still a challenge. Another technique that autocorrelation due to its collinear nature, from which the requires high spatial coherence for few-cycle pulses intensity autocorrelation can be calculated. The field measurement using an angular tunable bi-mirror for non- autocorrelation can also be measured, from which the optical pulse collinear autocorrelation is reported in ref. [11]. spectrum can be calculated. A theoretical model based on In this work, a MEMS-based autocorrelator that uses a Gaussian beam propagation is developed to study the effect of Michelson interferometer fabricated using silicon optical beam divergence, pulse dispersion, tilt angle between the interferometer mirrors, and amplitude mismatch between the micromachining technology is reported. -

Soliton Molecules and Multisoliton States in Ultrafast Fibre Lasers: Intrinsic Complexes in Dissipative Systems
applied sciences Review Soliton Molecules and Multisoliton States in Ultrafast Fibre Lasers: Intrinsic Complexes in Dissipative Systems Lili Gui 1,*, Pan Wang 2, Yihang Ding 2, Kangjun Zhao 2, Chengying Bao 3, Xiaosheng Xiao 2 and Changxi Yang 2,* 1 4th Physics Institute and Research Center SCoPE, University of Stuttgart, Pfaffenwaldring 57, 70569 Stuttgart, Germany 2 State Key Laboratory of Precision Measurement Technology and Instruments, Department of Precision Instruments, Tsinghua University, Beijing 100084, China; [email protected] (P.W.); [email protected] (Y.D.); [email protected] (K.Z.); [email protected] (X.X.) 3 Watson Laboratory of Applied Physics, California Institute of Technology, Pasadena, CA 91125, USA; [email protected] * Correspondence: [email protected] (L.G.); [email protected] (C.Y.); Tel.: +86-10-6277-2824 (C.Y.) Received: 14 December 2017; Accepted: 23 January 2018; Published: 29 January 2018 Featured Application: High-capacity telecommunication, advanced time-resolved spectroscopy, self-organization and chaos in dissipative systems. Abstract: Benefiting from ultrafast temporal resolution, broadband spectral bandwidth, as well as high peak power, passively mode-locked fibre lasers have attracted growing interest and exhibited great potential from fundamental sciences to industrial and military applications. As a nonlinear system containing complex interactions from gain, loss, nonlinearity, dispersion, etc., ultrafast fibre lasers deliver not only conventional single soliton but also soliton bunching with different types. In analogy to molecules consisting of several atoms in chemistry, soliton molecules (in other words, bound solitons) in fibre lasers are of vital importance for in-depth understanding of the nonlinear interaction mechanism and further exploration for high-capacity fibre-optic communications. -

Multi-Purpose Device for Analyzing and Measuring Ultra-Short Pulses
University of Central Florida STARS Electronic Theses and Dissertations, 2004-2019 2016 Multi-Purpose device for analyzing and measuring ultra-short pulses Naman Anilkumar Mehta University of Central Florida Part of the Electromagnetics and Photonics Commons, and the Optics Commons Find similar works at: https://stars.library.ucf.edu/etd University of Central Florida Libraries http://library.ucf.edu This Masters Thesis (Open Access) is brought to you for free and open access by STARS. It has been accepted for inclusion in Electronic Theses and Dissertations, 2004-2019 by an authorized administrator of STARS. For more information, please contact [email protected]. STARS Citation Mehta, Naman Anilkumar, "Multi-Purpose device for analyzing and measuring ultra-short pulses" (2016). Electronic Theses and Dissertations, 2004-2019. 5135. https://stars.library.ucf.edu/etd/5135 MULTI-PURPOSE DEVICE FOR ANALYZING AND MEASURING ULTRA-SHORT PULSES by NAMAN ANILKUMAR MEHTA B.S. U.V. Patel College of Engineering, 2013 A thesis submitted in partial fulfilment of the requirements for the degree of Master of Science in the Department of Engineering in the College of Optics and Photonics: CREOL & FPCE at the University of Central Florida Orlando, Florida Summer Term 2016 Major Professor: Axel Schulzgen c 2016 Naman Anilkumar Mehta ii ABSTRACT Intensity auto correlator is device to measure pulse widths of ultrashort pulses on the order of pi- coseconds and femtoseconds. I have built an in-house, compact, portable, industry standard inten- sity auto correlator for measuring ultrashort pulse-widths. My device is suitable for pulse-widths from 500 ps to 50 fs. The impetus for developing this instrument stemmed from our development of a multicore-fiber laser for high power laser applications, which also produces very short pulses that cannot be measured with an oscilloscope. -

High Power Mode-Locked Semiconductor Lasers and Their Applications
University of Central Florida STARS Electronic Theses and Dissertations, 2004-2019 2008 High Power Mode-locked Semiconductor Lasers And Their Applications Shinwook Lee University of Central Florida Part of the Electromagnetics and Photonics Commons, and the Optics Commons Find similar works at: https://stars.library.ucf.edu/etd University of Central Florida Libraries http://library.ucf.edu This Doctoral Dissertation (Open Access) is brought to you for free and open access by STARS. It has been accepted for inclusion in Electronic Theses and Dissertations, 2004-2019 by an authorized administrator of STARS. For more information, please contact [email protected]. STARS Citation Lee, Shinwook, "High Power Mode-locked Semiconductor Lasers And Their Applications" (2008). Electronic Theses and Dissertations, 2004-2019. 3722. https://stars.library.ucf.edu/etd/3722 HIGH POWER MODE-LOCKED SEMICONDUCTOR LASERS AND THEIR APPLICATIONS by SHINWOOK LEE B.Sc. Sogang University, Republic of Korea, 1993 M.Sc. Sogang University, Republic of Korea, 1995 A dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the College of Optics and Photonics/CREOL at the University of Central Florida Orlando, Florida Spring Term 2008 Major Professor: Peter J. Delfyett, Jr. © 2008 Shinwook Lee ii For My Mother and Wife. iii ABSTRACT In this dissertation, a novel semiconductor mode-locked oscillator which is an extension of eXtreme Chirped Pulse Amplification (XCPA) is investigated. An eXtreme Chirped Pulse Oscillator (XCPO) implemented with a Theta cavity also based on a semiconductor gain is presented for generating more than 30ns frequency-swept pulses with more than 100pJ of pulse energy and 3.6ps compressed pulses directly from the oscillator. -

Triple-Optical Autocorrelation for Direct Optical Pulse-Shape Measurement
APPLIED PHYSICS LETTERS VOLUME 81, NUMBER 8 19 AUGUST 2002 Triple-optical autocorrelation for direct optical pulse-shape measurement Tzu-Ming Liu, Yin-Chieh Huang, Gia-Wei Chern, Kung-Hsuan Lin, Chih-Jie Lee, Yu-Chueh Hung, and Chi-Kuang Suna) Department of Electrical Engineering and Graduate Institute of Electro-Optical Engineering, National Taiwan University, Taipei 10617, Taiwan, Republic of China ͑Received 4 September 2001; accepted for publication 24 June 2002͒ Triple optical autocorrelation of femtosecond optical pulses was realized simply with third-harmonic-generation technique. This optical technique provides complete knowledge of transient pulse intensity variation directly in time domain. Only analytic calculation is needed to obtain the pulse-shape from data without direction-of-time ambiguity. Combined with a spectral measurement and the Gerchberg–Saxton algorithm, except for pulses with complete temporal and spectral symmetry that will cause a twofold ambiguity, exact phase variation in time can also be retrieved through an iterative calculation with an O(n) complexity. © 2002 American Institute of Physics. ͓DOI: 10.1063/1.1501453͔ In most time domain applications and studies, measuring only. In previous studies, it had been shown that a triple the ultrashort optical pulse-shape is a crucial diagnosis. Be- correlation function is sufficient to determine the temporal cause electronic devices are too slow to measure temporal intensity of a pulse with direct mathematical calculation.11,12 evolution of femtosecond optical pulses, it is thus important Here we realize this technique for direct optical pulse-shape to develop optical techniques based on slow electronic detec- measurement based on simply one stage of a THG effect. -

ACTIVE and PASSIVE MODE LOCKING of CONTINUOUSLY OPERATING RHODAMINE 6G Dye Lasers
NBSIR 73-347 ACTIVE AND PASSIVE MODE LOCKING OF CONTINUOUSLY OPERATING RHODAMINE 6G DYE LASERS Andre Scavennec Norris S. Nahman Electromagnetics Division Institute for Basic Standards National Bureau of Standards Boulder, Colorado 80302 February 1974 NBSIR 73-347 ACTIVE AND PASSIVE MODE LOCKING OF CONTINUOUSLY OPERATING RHODAMINE 6G DYE LASERS Andre Scavennec Norris S. Nahman Electromagnetics Division Institute for Basic Standards National Bureau of Standards Boulder, Colorado 80302 February 1974 U.S. DEPARTMENT OF COMMERCE, Frederick B. Dent, Secretary NATIONAL BUREAU OF STANDARDS Richard W Roberts Director NBSIR 73-347 ACTIVE AND PASSIVE MODE LOCKING OF CONTINUOUSLY OPERATING RHODAMINE 6G DYE LASERS Andre Scavennec Norris S. Nahman Electromagnetics Division Institute for Basic Standards National Bureau of Standards Boulder, Colorado 80302 February 1974 U.S. DEPARTMENT OF COMMERCE, Frederick B. Dent, Secretary NATIONAL BUREAU OF STANDARDS Richard W Roberts Director CONTENTS Ch apter Page 1. INTRODUCTION 2 2. THE CW Rh 6G LASER -- 3 3. MODE LOCKING IN A LASER 11 4. ACTIVE MODE LOCKING OF THE CW Rh 6G LASER 15 5. PASSIVE MODE LOCKING OF THE CW Rh 6G LASER---- 22 6. COMPARISON OF THE TWO TECHNIQUES 2 8 7. CONCLUSION 30 REFERENCES-- 34 LIST OF FIGURES AND TABLES Figure 2-1. Energy level diagram o£ Rh 6G 36 Figure 2-2. CW dye laser 37 Figure 2-3. Dye cells 38 Figure 2-4. Dye circulating system 39 Figure 3-1. Laser modes and mode locked output 40 Figure 4-1. Active mode locking, experimental setup-- 41 Figure 4-2. Acoustooptic modulator 42 Figure 4-3. Modulator driving circuit 43 Figure 4-4. -

Nanoscale Force Sensing of an Ultrafast Nonlinear Optical Response
Nanoscale force sensing of an ultrafast nonlinear optical response Zeno Schumachera,1,2 , Rasa Rejalia, Raphael Pachlatkoa , Andreas Spielhofera, Philipp Naglerb, Yoichi Miyaharaa, David G. Cookea , and Peter Grutter¨ a,1 aDepartment of Physics, McGill University, Montreal QC H3A 2T8, Canada; and bDepartment of Physics, University of Regensburg, 93053 Regensburg, Germany Edited by Jianming Cao, Florida State University, Tallahassee, FL, and accepted by Editorial Board Member Zachary Fisk July 2, 2020 (received for review March 2, 2020) The nonlinear optical response of a material is a sensitive probe Results of electronic and structural dynamics under strong light fields. We split the output of a mode-locked femtosecond fiber laser The induced microscopic polarizations are usually detected via operating at 80-MHz repetition rate and 780-nm central wave- their far-field light emission, thus limiting spatial resolution. Sev- length, 200 mW (Toptica FemtoFiber pro NIR), to generate eral powerful near-field techniques circumvent this limitation two coherent pulse trains, with a well-defined temporal delay by employing local nanoscale scatterers; however, their signal between the two. The pulse trains can be arranged in a non- strength scales unfavorably as the probe volume decreases. Here, collinear or collinear fashion and focused onto the tip–sample we demonstrate that time-resolved atomic force microscopy is junction of an AFM incorporated in a UHV system. As the two capable of temporally and spatially resolving the microscopic, pulses are delayed with respect to each other, the light inten- electrostatic forces arising from a nonlinear optical polarization sity at the tip–sample junction oscillates, in delay time, with the in an insulating dielectric driven by femtosecond optical fields. -

Jannik Ehlert Characterization Frequency-Resolved Optical Gating
Frequency-resolved optical gating for broadband pulse characterization Jannik Ehlert Supervisors: Prof. dr. ir. Roel Baets, Prof. Valdas Pasiskevicius (KTH) Master's dissertation submitted in order to obtain the academic degree of European Master of Science in Photonics Department of Information Technology Chair: Prof. dr. ir. Bart Dhoedt Faculty of Engineering and Architecture Academic year 2016-2017 Frequency-resolved optical gating for broadband pulse characterization Jannik Ehlert Supervisors: Prof. dr. ir. Roel Baets, Prof. Valdas Pasiskevicius (KTH) Master's dissertation submitted in order to obtain the academic degree of European Master of Science in Photonics Department of Information Technology Chair: Prof. dr. ir. Bart Dhoedt Faculty of Engineering and Architecture Academic year 2016-2017 Foreword This thesis worth 30 credit points in the field of photonics is my final piece inthe puzzle of my Master’s studies. It describes the experimental work of building a pulse characterization setup at KTH – Royal Institute of Technology in Stockholm. The Laser physics group of KTH has been my main reason to spend the second year of my Master’s studies in Sweden. Before starting lessons at KTH, I had been loaded with a good basis of theoretical knowledge from one semester each at both Ghent University and Vrije Universiteit Brussel. When starting the experimental work I first felt I was limited in apply- ing my knowledge and did not know when to ask once I was stuck, but step by step I could broaden my skills in many areas and feel very satisfied with the result. Therefore, I would like to thank several individuals and groups who have helped and supported me along the way.