Inhibition of Phosphodiesterase 4 Modulates Cytokine Induction From
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Download Product Insert (PDF)
PRODUCT INFORMATION Piclamilast Item No. 29170 CAS Registry No.: 144035-83-6 Formal Name: 3-(cyclopentyloxy)-N-(3,5-dichloro- 4-pyridinyl)-4-methoxy-benzamide O Synonyms: RP 73401, RPR 73401 Cl H MF: C18H18Cl2N2O3 FW: 381.3 N O Purity: ≥98% O Supplied as: A solid N Storage: -20°C Cl Stability: ≥2 years Information represents the product specifications. Batch specific analytical results are provided on each certificate of analysis. Laboratory Procedures Piclamilast is supplied as a solid. A stock solution may be made by dissolving the piclamilast in the solvent of choice, which should be purged with an inert gas. Piclamilast is soluble in organic solvents such as ethanol and DMSO. The solubility of piclamilast in ethanol is approximately 20 mM and approximately 100 mM in DMSO. Description 1 Piclamilast is a phosphodiesterase 4 (PDE4) inhibitor (IC50 = 0.31 nM). It is selective for PDE4 over PDE1, 2,3 PDE2, PDE3, PDE5, and PDE7A in cell-free assays (IC50s = >100, 40, >100, 14, and >10 μM, respectively). Piclamilast inhibits superoxide production by guinea pig eosinophils and histamine-induced contraction 1,4 in isolated guinea pig trachea (IC50s = 24 and 2 nM, respectively). It inhibits eosinophil, neutrophil, lymphocyte, and TNF-α accumulation in bronchoalveolar lavage fluid (BALF) from ovalbumin-sensitized 5 rats (ED50s = 23.8, 14.1, 19.5, and 14.4 μmol/kg, respectively). Piclamilast also inhibits ovalbumin-induced 2 bronchoconstriction in a guinea pig model of asthma (ED50 = 0.033 mg/kg). References 1. Ashton, M.J., Cook, D.C., Fenton, G., et al. Selective type IV phosphodiesterase inhibitors as antiasthmatic agents. -
![[3H]-Piclamilast and [3H]-Rolipram](https://docslib.b-cdn.net/cover/0090/3h-piclamilast-and-3h-rolipram-100090.webp)
[3H]-Piclamilast and [3H]-Rolipram
JPET Fast Forward. Published on January 24, 2003 as DOI: 10.1124/jpet.102.047407 JPET FastThis articleForward. has not Published been copyedited on and January formatted. 24,The final2003 version as DOI:10.1124/jpet.102.047407 may differ from this version. Inhibitor Binding to Type 4 Phosphodiesterase (PDE4) Assessed Using [3H]-Piclamilast and [3H]-Rolipram Yu Zhao, Han-Ting Zhang, and James M. O’Donnell Department of Pharmacology University of Tennessee Health Science Center Downloaded from Memphis, Tennessee jpet.aspetjournals.org at ASPET Journals on September 27, 2021 1 Copyright 2003 by the American Society for Pharmacology and Experimental Therapeutics. JPET Fast Forward. Published on January 24, 2003 as DOI: 10.1124/jpet.102.047407 This article has not been copyedited and formatted. The final version may differ from this version. Running title: Inhibitor binding to PDE4 Correspondence should be addressed to: James M. O’Donnell, Ph.D. Department of Pharmacology University of Tennessee Health Science Center 874 Union Avenue Downloaded from Memphis, TN 38163 jpet.aspetjournals.org Phone: 901-448-3621 Fax: 901-448-3849 Email: [email protected] at ASPET Journals on September 27, 2021 Number of text page: 31 Number of tables: 3 Number of figures: 7 Number of references: 50 Number of words: Abstract (241); Introduction (716); Discussion (1544) Abbreviations: EHNA, erythro-9-(2-hydroxy-3-nonyl)adenine; HARBS, high-affinity rolipram binding site; LARBS, low-affinity rolipram binding site; IBMX, 3-isobutyl-1- methylxanthine; PDE, phosphodiesterase Section: Neuropharmacology 2 JPET Fast Forward. Published on January 24, 2003 as DOI: 10.1124/jpet.102.047407 This article has not been copyedited and formatted. -

Chapter Introduction
VU Research Portal Trypanosoma brucei phosphodiesterase B1 as a drug target for Human African Trypanosomiasis Jansen, C.J.W. 2015 document version Publisher's PDF, also known as Version of record Link to publication in VU Research Portal citation for published version (APA) Jansen, C. J. W. (2015). Trypanosoma brucei phosphodiesterase B1 as a drug target for Human African Trypanosomiasis. General rights Copyright and moral rights for the publications made accessible in the public portal are retained by the authors and/or other copyright owners and it is a condition of accessing publications that users recognise and abide by the legal requirements associated with these rights. • Users may download and print one copy of any publication from the public portal for the purpose of private study or research. • You may not further distribute the material or use it for any profit-making activity or commercial gain • You may freely distribute the URL identifying the publication in the public portal ? Take down policy If you believe that this document breaches copyright please contact us providing details, and we will remove access to the work immediately and investigate your claim. E-mail address: [email protected] Download date: 05. Oct. 2021 An introduction into phosphodiesterases and their potential role as drug targets for neglected diseases Chapter 1 4 CHAPTER 1 1.1 Human African Trypanosomiasis Human African Trypanomiasis (HAT), also known as African sleeping sickness, is a deadly infectious disease caused by the kinetoplastid Trypanosoma -

Stems for Nonproprietary Drug Names
USAN STEM LIST STEM DEFINITION EXAMPLES -abine (see -arabine, -citabine) -ac anti-inflammatory agents (acetic acid derivatives) bromfenac dexpemedolac -acetam (see -racetam) -adol or analgesics (mixed opiate receptor agonists/ tazadolene -adol- antagonists) spiradolene levonantradol -adox antibacterials (quinoline dioxide derivatives) carbadox -afenone antiarrhythmics (propafenone derivatives) alprafenone diprafenonex -afil PDE5 inhibitors tadalafil -aj- antiarrhythmics (ajmaline derivatives) lorajmine -aldrate antacid aluminum salts magaldrate -algron alpha1 - and alpha2 - adrenoreceptor agonists dabuzalgron -alol combined alpha and beta blockers labetalol medroxalol -amidis antimyloidotics tafamidis -amivir (see -vir) -ampa ionotropic non-NMDA glutamate receptors (AMPA and/or KA receptors) subgroup: -ampanel antagonists becampanel -ampator modulators forampator -anib angiogenesis inhibitors pegaptanib cediranib 1 subgroup: -siranib siRNA bevasiranib -andr- androgens nandrolone -anserin serotonin 5-HT2 receptor antagonists altanserin tropanserin adatanserin -antel anthelmintics (undefined group) carbantel subgroup: -quantel 2-deoxoparaherquamide A derivatives derquantel -antrone antineoplastics; anthraquinone derivatives pixantrone -apsel P-selectin antagonists torapsel -arabine antineoplastics (arabinofuranosyl derivatives) fazarabine fludarabine aril-, -aril, -aril- antiviral (arildone derivatives) pleconaril arildone fosarilate -arit antirheumatics (lobenzarit type) lobenzarit clobuzarit -arol anticoagulants (dicumarol type) dicumarol -

Phosphodiesterase (PDE)
Phosphodiesterase (PDE) Phosphodiesterase (PDE) is any enzyme that breaks a phosphodiester bond. Usually, people speaking of phosphodiesterase are referring to cyclic nucleotide phosphodiesterases, which have great clinical significance and are described below. However, there are many other families of phosphodiesterases, including phospholipases C and D, autotaxin, sphingomyelin phosphodiesterase, DNases, RNases, and restriction endonucleases, as well as numerous less-well-characterized small-molecule phosphodiesterases. The cyclic nucleotide phosphodiesterases comprise a group of enzymes that degrade the phosphodiester bond in the second messenger molecules cAMP and cGMP. They regulate the localization, duration, and amplitude of cyclic nucleotide signaling within subcellular domains. PDEs are therefore important regulators ofsignal transduction mediated by these second messenger molecules. www.MedChemExpress.com 1 Phosphodiesterase (PDE) Inhibitors, Activators & Modulators (+)-Medioresinol Di-O-β-D-glucopyranoside (R)-(-)-Rolipram Cat. No.: HY-N8209 ((R)-Rolipram; (-)-Rolipram) Cat. No.: HY-16900A (+)-Medioresinol Di-O-β-D-glucopyranoside is a (R)-(-)-Rolipram is the R-enantiomer of Rolipram. lignan glucoside with strong inhibitory activity Rolipram is a selective inhibitor of of 3', 5'-cyclic monophosphate (cyclic AMP) phosphodiesterases PDE4 with IC50 of 3 nM, 130 nM phosphodiesterase. and 240 nM for PDE4A, PDE4B, and PDE4D, respectively. Purity: >98% Purity: 99.91% Clinical Data: No Development Reported Clinical Data: No Development Reported Size: 1 mg, 5 mg Size: 10 mM × 1 mL, 10 mg, 50 mg (R)-DNMDP (S)-(+)-Rolipram Cat. No.: HY-122751 ((+)-Rolipram; (S)-Rolipram) Cat. No.: HY-B0392 (R)-DNMDP is a potent and selective cancer cell (S)-(+)-Rolipram ((+)-Rolipram) is a cyclic cytotoxic agent. (R)-DNMDP, the R-form of DNMDP, AMP(cAMP)-specific phosphodiesterase (PDE) binds PDE3A directly. -

The Use of Stems in the Selection of International Nonproprietary Names (INN) for Pharmaceutical Substances
WHO/PSM/QSM/2006.3 The use of stems in the selection of International Nonproprietary Names (INN) for pharmaceutical substances 2006 Programme on International Nonproprietary Names (INN) Quality Assurance and Safety: Medicines Medicines Policy and Standards The use of stems in the selection of International Nonproprietary Names (INN) for pharmaceutical substances FORMER DOCUMENT NUMBER: WHO/PHARM S/NOM 15 © World Health Organization 2006 All rights reserved. Publications of the World Health Organization can be obtained from WHO Press, World Health Organization, 20 Avenue Appia, 1211 Geneva 27, Switzerland (tel.: +41 22 791 3264; fax: +41 22 791 4857; e-mail: [email protected]). Requests for permission to reproduce or translate WHO publications – whether for sale or for noncommercial distribution – should be addressed to WHO Press, at the above address (fax: +41 22 791 4806; e-mail: [email protected]). The designations employed and the presentation of the material in this publication do not imply the expression of any opinion whatsoever on the part of the World Health Organization concerning the legal status of any country, territory, city or area or of its authorities, or concerning the delimitation of its frontiers or boundaries. Dotted lines on maps represent approximate border lines for which there may not yet be full agreement. The mention of specific companies or of certain manufacturers’ products does not imply that they are endorsed or recommended by the World Health Organization in preference to others of a similar nature that are not mentioned. Errors and omissions excepted, the names of proprietary products are distinguished by initial capital letters. -

Suplementum 12/1 MAKETA 7/1 2/23/12 12:12 PM Stránka 16
suplementum 12/1 _MAKETA 7/1 2/23/12 12:12 PM Stránka 16 16 ACTA MEDICA MARTINIANA 2012 Suppl. 1 DOI: 10.2478/v10201-011-0023-7 SELECTIVE INHIBITION OF PHOSPHODIESTERASE 7 (PDE7) BY BRL50481 IN HEALTHY AND OVALBUMIN-SENSITIZED GUINEA PIGS Christensen I.1, Miskovicova H. 1, Porvaznik I.1,JoskovaM.1, Mokra D.2, Mokry J.1 1Department of Pharmacology Jessenius Faculty of Medicine, Comenius University, Martin 2Department of Physiology, Jessenius Faculty of Medicine, Comenius University, Martin, Slovak Republic A b stract Phosphodiesterase (PDE) inhibitors may have significant clinical benefit in respiratory diseases associated with inflammation. The aim was to evaluate effects of selective PDE7 inhibitor (BRL50481) on citric acid induced cough, in vivo and in vitro airway smooth muscle reactivity in both healthy and ovalbumin sensitized guinea pigs, as well as its effectiveness in changes of blood cells count. Tested drugs were administered intraperitoneally to male guinea pigs once daily for 7 days either vehicle 10% DMSO (dimethyl sulfoxide) 3 ml/kg (as control) or BRL50481 1 mg/kg. Double chamber whole body plethysmo- graph was used for evaluation of citric acid (0.6 M) induced cough and specific airway resistance. Organ bath method was used for measurement of tracheal and lung tissue strips contractions evoked by cumulative doses (10- 8 – 10-3 mol/L) of acetylcholine (ACH) and histamine (HIS). In healthy guinea pigs we did not observe significant effect of tested drug BRL50481 on in vitro contractions (sim- ilarly to in vivo conditions). The effect on cough was in healthy animals negligible. In ovalbumin-sensitized ani- mals, more pronounced in vitro relaxing effect of BRL50481 in HIS induced contractions was observed with simi- lar results in vivo and no significant change in number of cough efforts. -

New Therapeutic Options in the Management of COPD – Focus on Roflumilast
International Journal of COPD Dovepress open access to scientific and medical research Open Access Full Text Article REVIEW New therapeutic options in the management of COPD – focus on roflumilast Sabina Antonela Antoniu Abstract: In chronic obstructive pulmonary disease (COPD) the inflammation occurring in the University of Medicine and Pharmacy, airways and in other lung tissues is complex and is orchestrated by various mediators including the Pulmonary Disease Division, isoenzyme 4 of the phosphodiesterases family (PDE4), which contributes to bronchoconstriction Pulmonary Disease University and inflammation. Various PDE4 inhibitors have been evaluated as potential therapies in asthma Hospital, Iasi, Romania or COPD but among these only roflumilast have been authorized in Europe to be used in patients with severe COPD as an add-on to the bronchodilator therapy. This review discusses the existing preclinical and clinical data supporting the use of roflumilast for this therapeutic indication and tackles some of the pending issues related to PDE4 in general and to roflumilast in particular. Keywords: COPD, roflumilast, PDE4, efficacy, safety Introduction COPD is an inflammatory disease of the airways related mainly to smoking and char- acterized by airflow limitation, which manifests clinically with dsypnea, cough, and sputum production, symptoms that aggravate disease severity and disease exacerbation. Chronic airways inflammation leads to their progressive obstruction, lung paren- chyma damage, and mucus hypersecretion. Subsequently these result in abnormalities in gas exchange at the pulmonary level and respiratory failure. Currently COPD is classified in 4 stages of severity depending on the degree of airways obstruction measured by FEV1 and with FEV1/FVC% ratio (Table 1). In COPD the therapeutic approach is aimed at treating disease exacerbation when this occurs and at slowing disease progression in the stable state. -
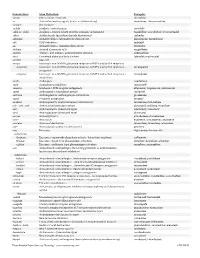
Downloaded from Survive Nursing | Survivenursing.Com V20110426
Generic Stem Stem Definition Examples -abine (see -arabine, -citabine) decitabine -ac Anti-inflammatory agents (acetic acid derivatives) bromfenac; dexpemedolac -acetam See -racetam -actide Synthetic corticotropins seractide -adol or -aldol- Analgesics (mixed opiate receptor agonists/ antagonists) tazadolene; spiradolene; levonantradol -adox Antibacterials (quinoline dioxide derivatives) carbadox -afenone Antiarrhythmics (propafenone derivatives) alprafenone; diprafenone -afil PDE5 inhibitors tadalafil -aj- Antiarrhythmics (ajmaline derivatives) lorajmine -aldrate Antacid aluminum salts magaldrate -algron Alpha1 - and alpha2 - adrenoreceptor agonists dabuzalgron -alol Combined alpha and beta blockers labetalol; medroxalol -amivir (see -vir) -ampa Ionotropic non-NMDA glutamate receptors (AMPA and/or KA receptors) -ampanel Ionotropic non-NMDA glutamate receptors (AMPA and/or KA receptors) ; becampanel antagonists -ampator Ionotropic non-NMDA glutamate receptors (AMPA and/or KA receptors) ; forampator modulators -andr- Androgens nandrolone -anib Angiogenesis inhibitors semaxanib -anserin Serotonin 5-HT2 receptor antagonists altanserin; tropanserin; adatanserin -antel Anthelmintics (undefined group) carbantel -antrone Antineoplastics; anthraquinone derivatives pixantrone -apsel P-selectin antagonists torapsel -arabine Antineoplastics (arabinofuranosyl derivatives) fazarabine; fludarabine aril-, -aril, -aril- Antiviral (arildone derivatives) pleconaril; arildone; fosarilate -arit Antirheumatics (lobenzarit type) lobenzarit; clobuzarit -arol -

Selective Phosphodiesterase 4 Inhibitors
Journal of Scientific & Indu strial Research Vol. 62, June 2003, pp 537-553 Selective Phosphodiesterase 4 Inhibitors - Emerging Trends in Astluna Therapy (Antiasthmatics-3) Ranju Gupta*, Oeepika Gandhi and Oharam Paul Jindal University In stitute of Pharmaceuti cal Sciences, Panjab Universit y, Chandigarh 160014 Considerable interest has been generated in th e potential utility of isozyme selective inhibitors of phosph odiesterases in the treatment of asthma and other inflali1matory disorders. Heterogeneity in ti ssue distribution as well as their different fun cti onal roles make these enzymes very attractive targets for medicinal chemi sts. To date at least II different fami lies of POE isozymes are known, among which POE 4 plays a major rol e in mod ul ating the activity of virtu ally all cell s involved in the inflammatory process. In hibitors of thi s enzyme family display impressive antiasthmatic ac ti vity by red ucing th e bronchial smooth mu scle tone and consid erable anti-inflammatory activity. The review details th e various classes of POE 4 inhibitors stru cturall y related to rolipram, nitraquazone and xa nthines, which appear to be very attractive models for synthesis of novel selecti ve POE 4 inhibitors potentially useful for the treatment of asthma and chronic obstructive pulmonary di seases. Rationale for the use of POE4 inhibitors in th e treatment of asthma is also discussed. Key words: Phosphodiesterase 4 inhibitors, Asthma therapy, Antiasthmatics, Isozyme, Rol ipram, Nitraquazonc. Xanthincs. Introduction Phosphodiesterase Superfamily There has been growing interest in recent years At least e leven different fami li es of POE tn the utility of selective phosphodiesterase (POE) isozymes are known based on the ir substrate inhibitors as novel targets for drug d iscovery. -

1 Elevated Intracellular Camp Concentration Mediates Growth
Elevated intracellular cAMP concentration mediates growth suppression in glioma cells Dewi Safitri1,2, Matthew Harris1, Harriet Potter1, Ho Yan Yeung1, Ian Winfield1, Liliya Kopanitsa3, Fredrik Svensson3,4, Taufiq Rahman1, Matthew T Harper1, David Bailey3, Graham Ladds1, *. 1 Department of Pharmacology, University of Cambridge, Tennis Court Road, Cambridge CB2 1PD, United Kingdom 2 Pharmacology and Clinical Pharmacy Research Group, School of Pharmacy, Bandung Institute of Technology, Bandung 40132, Indonesia 3 IOTA Pharmaceuticals Ltd, Cambridge University Biomedical Innovation Hub, Clifford Allbutt Building, Hills Road, Cambridge CB2 0AH, United Kingdom 4 Computational Medicinal Chemistry, Alzheimer’s Research UK UCL Drug Discovery Institute, The Cruciform Building, Gower Street, London, WC1E 6BT *Corresponding author: Dr Graham Ladds, Department of Pharmacology, University of Cambridge, Tennis Court Road, Cambridge, CB2 1PD Tel; +44 (0) 1223 334020. Email: [email protected]. ABSTRACT Supressed levels of intracellular cAMP have been associated with malignancy. Thus, elevating cAMP through activation of adenylyl cyclase (AC) or by inhibition of phosphodiesterase (PDE) may be therapeutically beneficial. Here, we demonstrate that elevated cAMP levels suppress growth in C6 cells (a model of glioma) through treatment with forskolin, an AC activator, or a range of small molecule PDE inhibitors 1 with differing selectivity profiles. Forskolin suppressed cell growth in a PKA-dependent manner by inducing a G2/M phase cell cycle arrest. In contrast, trequinsin (a non- selective PDE2/3/7 inhibitor), not only inhibited cell growth via PKA, but also stimulated (independent of PKA) caspase-3/-7 and induced an aneuploidy phenotype. Interestingly, a cocktail of individual PDE 2,3,7 inhibitors suppressed cell growth in a manner analogous to forskolin but not trequinsin. -

Stembook 2018.Pdf
The use of stems in the selection of International Nonproprietary Names (INN) for pharmaceutical substances FORMER DOCUMENT NUMBER: WHO/PHARM S/NOM 15 WHO/EMP/RHT/TSN/2018.1 © World Health Organization 2018 Some rights reserved. This work is available under the Creative Commons Attribution-NonCommercial-ShareAlike 3.0 IGO licence (CC BY-NC-SA 3.0 IGO; https://creativecommons.org/licenses/by-nc-sa/3.0/igo). Under the terms of this licence, you may copy, redistribute and adapt the work for non-commercial purposes, provided the work is appropriately cited, as indicated below. In any use of this work, there should be no suggestion that WHO endorses any specific organization, products or services. The use of the WHO logo is not permitted. If you adapt the work, then you must license your work under the same or equivalent Creative Commons licence. If you create a translation of this work, you should add the following disclaimer along with the suggested citation: “This translation was not created by the World Health Organization (WHO). WHO is not responsible for the content or accuracy of this translation. The original English edition shall be the binding and authentic edition”. Any mediation relating to disputes arising under the licence shall be conducted in accordance with the mediation rules of the World Intellectual Property Organization. Suggested citation. The use of stems in the selection of International Nonproprietary Names (INN) for pharmaceutical substances. Geneva: World Health Organization; 2018 (WHO/EMP/RHT/TSN/2018.1). Licence: CC BY-NC-SA 3.0 IGO. Cataloguing-in-Publication (CIP) data.