The Development of Novel Biocatalysts for the Asymmetric Hydroamination of Alkenes
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Mechanistic Aspects of the Reaction of Oxime Tosylates with Grignard Reagents
University of New Hampshire University of New Hampshire Scholars' Repository Doctoral Dissertations Student Scholarship Spring 1965 MECHANISTIC ASPECTS OF THE REACTION OF OXIME TOSYLATES WITH GRIGNARD REAGENTS STANFORD SALAVATORE PELOSI JR. Follow this and additional works at: https://scholars.unh.edu/dissertation Recommended Citation PELOSI, STANFORD SALAVATORE JR., "MECHANISTIC ASPECTS OF THE REACTION OF OXIME TOSYLATES WITH GRIGNARD REAGENTS" (1965). Doctoral Dissertations. 811. https://scholars.unh.edu/dissertation/811 This Dissertation is brought to you for free and open access by the Student Scholarship at University of New Hampshire Scholars' Repository. It has been accepted for inclusion in Doctoral Dissertations by an authorized administrator of University of New Hampshire Scholars' Repository. For more information, please contact [email protected]. This dissertation has been 65-9084 microfilmed exactly as received PELOSI, Jr., Stanford Salvatore, 1938- MECHANISTIC ASPECTS OF THE REACTION OF OXIME TOSYLATES WITH GRIGNARD REAGENTS. i University of New Hampshire, Ph.D., 1965 ; Chemistry, organic i I i University Microfilms, Inc., Ann Arbor, Michigan i Reproduced with permission of the copyright owner. Further reproduction prohibited without permission. MECHANISTIC ASPECTS OF THE REACTION OF OXIME TOSYLATES WITH GRIGNARD REAGENTS BY STANFORD SALVATORE PELOSI, JR. B. S., Boston College, 1960 A THESIS Submitted to the University of New Hampshire, In Partial Fulfillment of The Requirements for the Degree of Doctor of Philosophy Graduate School Department of Chemistry March, 1965 Reproduced with permission of the copyright owner. Further reproduction prohibited without permission. This thesis has been examined .and approved. I | :.c < I ' l Y . 7 1 / 1 / Ctrvi HS Dat Reproduced with permission of the copyright owner. -

Enhanced Isotopic Ratio Outlier Analysis (IROA) Peak Detection and Identification with Ultra-High Resolution GC-Orbitrap/MS
H OH metabolites OH Article Enhanced Isotopic Ratio Outlier Analysis (IROA) Peak Detection and Identification with Ultra-High Resolution GC-Orbitrap/MS: Potential Application for Investigation of Model Organism Metabolomes Yunping Qiu 1 ID , Robyn D. Moir 2, Ian M. Willis 2, Suresh Seethapathy 3, Robert C. Biniakewitz 3 and Irwin J. Kurland 1,* 1 Stable Isotope and Metabolomics Core Facility, Diabetes Center, Department of Medicine, Albert Einstein College of Medicine, Bronx, NY 10461, USA; [email protected] 2 Department of Biochemistry, Albert Einstein College of Medicine, Bronx, NY 10461, USA; [email protected] (R.D.M.); [email protected] (I.M.W.) 3 Thermo Fisher Scientific, Somerset, NJ 08873, USA; suresh.seethapathy@thermofisher.com (S.S.); [email protected] (R.C.B.) * Correspondence: [email protected]; Tel.: +1-718-678-1091 Received: 7 November 2017; Accepted: 10 January 2018; Published: 18 January 2018 Abstract: Identifying non-annotated peaks may have a significant impact on the understanding of biological systems. In silico methodologies have focused on ESI LC/MS/MS for identifying non-annotated MS peaks. In this study, we employed in silico methodology to develop an Isotopic Ratio Outlier Analysis (IROA) workflow using enhanced mass spectrometric data acquired with the ultra-high resolution GC-Orbitrap/MS to determine the identity of non-annotated metabolites. The higher resolution of the GC-Orbitrap/MS, together with its wide dynamic range, resulted in more IROA peak pairs detected, and increased reliability of chemical formulae generation (CFG). IROA uses two different 13C-enriched carbon sources (randomized 95% 12C and 95% 13C) to produce mirror image isotopologue pairs, whose mass difference reveals the carbon chain length (n), which aids in the identification of endogenous metabolites. -

Important Features of the Potential Energy Surface of the Methylamine Plus O(1D) Reaction
Physical Chemistry Chemical Physics Important Features of the Potential Energy Surface of the Methylamine Plus O(1D) Reaction Journal: Physical Chemistry Chemical Physics Manuscript ID CP-ART-09-2019-005039.R1 Article Type: Paper Date Submitted by the 17-Oct-2019 Author: Complete List of Authors: Wolf, Mark; University of Georgia , Center for Computational Quantum Chemsitry; Hoobler, Preston; Covenant College, Chemistry; University of Georgia Franklin College of Arts and Sciences, Center for Computational Quantum Chemistry Turney, Justin; University of Georgia, Center for Computational Chemistry Schaefer, Henry; University of Georgia, Computational Chemistry Page 1 of 13 Physical Chemistry Chemical Physics Journal Name Important Features of the Potential Energy Surface of the Methy- lamine Plus O(1D) Reaction † Mark E. Wolf, Preston R. Hoobler, Justin M. Turney, and Henry F. Schaefer III∗ This research presents an ab initio characterization of the potential energy surface for the methy- lamine plus 1D oxygen atom reaction, which may be relevant to interstellar chemistry. Ge- ometries and harmonic vibrational frequencies were determined for all stationary points at the CCSD(T)/aug-cc-pVTZ level of theory. The focal point method along with several additive correc- tions was used to obtain reliable CCSDT(Q)/CBS potential energy surface features. Extensive conformational analysis and intrinsic reaction coordinate computations were performed to ensure accurate chemical connectivity of the stationary points. Five minima were determined to be pos- sible products of this reaction and three novel transition states were found that were previously unreported or mislabeled in the literature. The pathways we present can be used to guide further searches for NH2 containing species in the interstellar medium. -

Inhibition of DNA Synthesis by Hydroxyurea: Structure-Activity Relationships1
[CANCER RESEARCH 27 Part 1, 535-540, March 1967] Inhibition of DNA Synthesis by Hydroxyurea: Structure-Activity Relationships1 CHARLES W. YOUNG, GERALD SCHOCHETMAN, SADIE HODAS, AND M. EARL BALIS Divisions of Clinical Chemothirapy and Biological Chemistry, Sloan-Ketlering Institute for Cancer Research, New York, New York 10021 SUMMARY allosteric effectors, altering substrate specificity and the rate of The following comix>unds inhibited incorporation of thy- the overall reaction (12, 13). The molecular mechanism by which midine-3H into the DNA of HeLa cells without significantly hydroxyurea inhibits deoxyribonucleotide synthesis is not known. impairing cellular incorjwration of uridine-3H into RNA or Substrate attack is unlikely because the drug does not impair loucine-3H into protein; the}' are listed in order of potency. synthesis of RNA; an attack ui>on the product of the reaction Dihydro.xyurea > jV-methylhydroxyurea > .V-acetylhydroxy- (deoxyribonucleotides) could not be demonstrated in ex|>eriments urea = hydroxyurea = A'-hydroxyguanidine = N-hydroxyure- with cell-free extracts (26). Because of the lack of structural thane = AT-ethylhydroxyurea > 3-phenyl-l-hydroxyurea = analogy between hydroxyurea and ribonucleotides or deoxyribo formamidoxime > Ar-mcthylacetohydroxamic acid = Ar-methyl- nucleotides, competition by the drug for the enzymatic substrate and/or allosteric-effector sites seems improbable. In an effort to hydroxylamine > acetohydroxamio acid. Hydroxylamine, JV-hydroxyglycine amide and 3-phenyl-l-hydroxy-2-thiourea clarify the relationship between molecular structure and inhibi tory activity, we have studied the effects of various hydroxyl- inhibited incori>oration of all three labeled precursors. Methoxy- amine, methoxyurea, and Ar-methylmethoxyurea had no effect amine and hydroxamic acid derivatives u)xjn protein and nucleic acid metabolism in HeLa cells. -

Assessing the Chemistry and Bioavailability of Dissolved Organic Matter from Glaciers and Rock 2 Glaciers 3
bioRxiv preprint doi: https://doi.org/10.1101/115808; this version posted April 11, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. 1 ASSESSING THE CHEMISTRY AND BIOAVAILABILITY OF DISSOLVED ORGANIC MATTER FROM GLACIERS AND ROCK 2 GLACIERS 3 4 1,2 1,3 3 1,4 5 Fegel, Timothy , Boot, Claudia M. , Broeckling, Corey D. , Hall, Ed K. 6 1. Natural Resource Ecology Laboratory, Colorado State University 2. U.S. Forest Service, 7 Rocky Mountain Research Station, 3. Department of Chemistry, Colorado State University 4. 8 Proteomics and Metabolomics Facility, Colorado State University 4. Department of Ecosystem 9 Science and Sustainability, Colorado State University 10 11 Tim Fegel [email protected] 12 13 14 Key Points: 15 Bioavailability of organic matter released from glaciers is greater than that of rock glaciers in the 16 Rocky Mountains. 17 The use of GC-MS for ecosystem metabolomics represents a novel approach for examining 18 complex organic matter pools. 19 Both glaciers and rock glaciers supply highly bioavailable sources of organic matter to alpine 20 headwaters in Colorado. 1 bioRxiv preprint doi: https://doi.org/10.1101/115808; this version posted April 11, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. -

Mechanic Aspects and Parameters for Biological Effect Monitoring
Erythrotoxicity of aliphatic hydroxylamines : mechanic aspects and parameters for biological effect monitoring Citation for published version (APA): Spooren, A. A. M. G. (2000). Erythrotoxicity of aliphatic hydroxylamines : mechanic aspects and parameters for biological effect monitoring. UM. https://doi.org/10.26481/dis.20001026as Document status and date: Published: 01/01/2000 DOI: 10.26481/dis.20001026as Document Version: Publisher's PDF, also known as Version of record Please check the document version of this publication: • A submitted manuscript is the version of the article upon submission and before peer-review. There can be important differences between the submitted version and the official published version of record. People interested in the research are advised to contact the author for the final version of the publication, or visit the DOI to the publisher's website. • The final author version and the galley proof are versions of the publication after peer review. • The final published version features the final layout of the paper including the volume, issue and page numbers. Link to publication General rights Copyright and moral rights for the publications made accessible in the public portal are retained by the authors and/or other copyright owners and it is a condition of accessing publications that users recognise and abide by the legal requirements associated with these rights. • Users may download and print one copy of any publication from the public portal for the purpose of private study or research. • You may not further distribute the material or use it for any profit-making activity or commercial gain • You may freely distribute the URL identifying the publication in the public portal. -

Methanamine, N-Methoxy
Methanamine, N-methoxy- Other names: CH3NHOCH3; Hydroxylamine, N,O-dimethyl-; Methoxyamine, N-methyl-; Methoxymethylamine; Methylamine, N-methoxy-; N,O-Dimethylhydroxylamine; N-Methoxy-N-methylamine; N-Methoxymethylamine; N-Methyl-O-methylhydroxylamine; N-Methylmethoxyamine; O,N-Dimethylhydroxylamine; O-Methyl-N-methylhydroxylamine. InChI: InChI=1S/C2H7NO/c1-3-4-2/h3H,1-2H3 InChI Key: KRKPYFLIYNGWTE-UHFFFAOYSA-N Formula: C2H7NO SMILES: CNOC Molecular Weight: 61.08 CAS: 1117-97-1 Physical Properties Property Value Unit Source ∆ G° -49.65 kJ/mol Joback Method f ∆ H° -163.36 Joback Method f gas kJ/mol ∆ H° 7.22 Joback Method fus kJ/mol ∆ H° 28.89 Joback Method vap kJ/mol IE 8.92 eV NIST Webbook logP -0.23 Crippen Method oct/wat P 5001.51 Joback Method c kPa T 317.75 Joback Method boil K T 488.59 Joback Method c K T 187.19 Joback Method fus K V 0.20 3 Joback Method c m /kg-mol Temperature Dependent Properties Property Value Unit Temperature (K) Source C 87.78 J/mol×K 317.75 Joback Method p,gas Property Value Unit Temperature (K) Source ∆ H 34.30 272.0 NIST Webbook vap kJ/mol Sources Joback Method: https://en.wikipedia.org/wiki/Joback_method NIST Webbook: http://webbook.nist.gov/cgi/inchi/InChI=1S/C2H7NO/c1-3-4-2/h3H,1-2H3 Crippen Method: http://pubs.acs.org/doi/abs/10.1021/ci990307l Legend C : Ideal gas heat capacity (J/mol×K). p,gas ∆ G°: Standard Gibbs free energy of formation (kJ/mol). f ∆ H° : Enthalpy of formation at standard conditions (kJ/mol). -

The Section of the Template to Be Replaced (E
Issue in Honor of Prof. William F. Bailey ARKIVOC 2011 (v) 230-262 Synthesis of amines by the electrophilic amination of organomagnesium, -zinc, -copper, and -lithium reagents Tahir Daşkapan Department of Chemistry, Ankara University Science Faculty, 06100 Beşevler, Ankara, Turkey E-mail: [email protected] Dedicated to Professor William F. Bailey on occasion of his 65th birthday DOI: http://dx.doi.org/10.3998/ark.5550190.0012.520 Contents 1. Introduction 2. Electrophilic Amination of Organomagnesium Reagents 3. Electrophilic Amination of Organozinc Reagents 4. Electrophilic Amination of Organocopper Reagents 5. Electrophilic Amination of Organolithium Reagents 6. Conclusions 7. Acknowledgement 8. References 1. Introduction Amines are important building blocks in the synthesis of various important organic compounds such as pharmaceuticals, agrochemicals, polymers, dyes, xerographic and photographic materials.1-5 Therefore, development of new synthetic methods for amines has attracted the attention of many researchers.6-11 Among modern amination methods, electrophilic amination of an easily available organometallic reagent has been found to be a viable alternative method and continues to be an active area for research in synthetic organic chemistry.12-16 To date various type of aminating reagents have been used in the electrophilic amination of organomagnesium, -zinc, -copper, and -lithium reagents for the introduction of free or protected amino moieties, such as haloamines, substituted hydroxylamines, imines, oxaziridines, oximes, diazonium salts, azodicarboxylates, azides, and metal amides. This review covers the literature published in the last twenty years in detail. In addition, older literature has been mentioned briefly. Page 230 ©ARKAT USA, Inc. Issue in Honor of Prof. William F. -
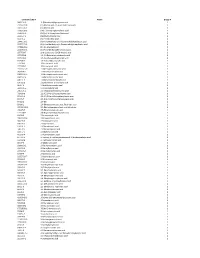
Chemical Classification List
Identifier/CAS # Name Group # 5683-31-8 3-(Trimethylsilyl)propynoic acid 1 21293-29-8 (+)-Abscisic acid, (2, 4-pentadienoic acid) 1 14375-45-2 (±)-Abscisic acid 1 35963-20-3 (1R)-(-)-10-Camphorsulfonic acid 1 3144-16-9 (1S)-(+)-10-Camphorsulfonic acid 1 2444-37-3 (Methylthio)acetic acid 1 611-71-2 (R)-(−)-Mandelic acid 1 20445-31-2 (R)-(+)-α-Methoxy-α-trifluoromethylphenylacetic acid 1 17257-71-5 (S)-(−)-α-Methoxy-α-(trifluoromethyl)phenylacetic acid 1 17199-29-0 (S )-(+)-Mandelic acid 1 26164-26-1 (S)-(+)-α-Methoxyphenylacetic acid 1 1077-28-7 ±)-α-Lipoic acid, DL-6,8-thioctic acid 1 1703-58-8 1,2,3,4-Butanetetracarboxylic acid 1 3971-31-1 1,3-Cyclohexanedicarboxylic acid 1 542-05-2 1,3-Acetonedicarboxylic acid 1 112-38-9 10-Undecenoic acid 1 2777-65-3 10-Undecynoic acid 1 71310-21-9 11-Mercaptoundecanoic acid 1 2834-05-1 11-Bromoundecanoic acid 1 69839-68-5 16-Mercaptohexadecanoic acid 1 4942-47-6 1-Adamantaneacetic acid 1 828-51-3 1-Adamantanecarboxylic acid 1 636-82-8 1-Cyclohexene-1-carboxylic acid 1 86-87-3 1-Naphthaleneacetic acid 1 3443-45-6 1-Pyrenebutyric acid 1 2062-25-1 2-(Trifluoromethyl)cinnamic acid 1 719-60-8 2,3,4,5,6-Pentafluorocinnamic acid 1 653-21-4 2,3,4,5,6-Pentafluorophenylacetic acid 1 94-75-7 2,4-D (2,4-Dichlorophenoxyacetic acid) 1 94-82-6 2,4-DB 1 89-86-1 2,4-Dihydroxybenzoic acid, Resorcylic acid 1 207234-00-2 2,4-Dinitrobenzenesulfonic acid dihydrate 1 490-79-9 2,5-Dihydroxybenzoic acid 1 1141-38-4 2,6-Naphthalenedicarboxylic acid 1 80-58-0 2-Bromobutyric acid 1 70610-87-6 2-Bromooctanoic acid 1 584-93-0 -

United States Patent Office Patented Jan
3,230,260 United States Patent Office Patented Jan. 18, 1966 2 3,230,260 acid this does not occur. Thus the relatively non-volatile PROCESS FOR THE SOLATION OF N,O-DIMETH acid salts of N,O-dimethylhydroxylamine can be formed YLHYDROXYLAMINE Such as the hydrochloride, the sulfate or hydrogen sul Jack A. Snyder, Claymont, Del, assignor to E. I. du Pont fate. Sulfamic acid or phosphoric acid can similarly be de Nemours and Company, Wilmington, Del, a cor 5 used though they are less preferred. An amount of acid poration of Delaware can be used which is at least stoichiometrically equivalent No Drawing. Filed May 26, 1961, Ser. No. 112,772 to N.O-dimethylhydroxylamine and is preferred. Less 2 Claims. (C. 260-583) than stoichiometric amounts can be used though as less This invention relates to the preparation of N.O-di and less acid is used there will be loss of yield. While methylhydroxylamine. The invention is more particu O the processes of the invention can be operated with a pH larly directed to processes for the isolation of NO up to about 9, it is ordinarily preferred to use a pH dimethylhydroxylamine from a mixture of O-methylhy around 1 or 2. droxylamine and N,O-dimethylhydroxylamine by adding The amount of O-methylhydroxylamine and N.O-di formaldehyde thereby to convert O-methylhydroxylamine methylhydroxylamine dissolved in an aqueous solution to O-methylformaldoxime which escapes as a gas. 5 will ordinarily be of the order of 1 molar though it can By the methylation of hydroxyurethane followed by be higher or lower.