A Conserved Role for the ALS-Linked Splicing Factor SFPQ in Repression of Pathogenic Cryptic Last Exons ✉ ✉ Patricia M
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Miasdb: a Database of Molecular Interactions Associated with Alternative Splicing of Human Pre-Mrnas
RESEARCH ARTICLE MiasDB: A Database of Molecular Interactions Associated with Alternative Splicing of Human Pre-mRNAs Yongqiang Xing1, Xiujuan Zhao1, Tao Yu2, Dong Liang1, Jun Li1, Guanyun Wei1, Guoqing Liu1, Xiangjun Cui1, Hongyu Zhao1, Lu Cai1* 1 School of Life Science and Technology, Inner Mongolia University of Science and Technology, Baotou, 014010, China, 2 School of Science, Inner Mongolia University of Science and Technology, Baotou, 014010, China a11111 * [email protected] Abstract Alternative splicing (AS) is pervasive in human multi-exon genes and is a major contributor to expansion of the transcriptome and proteome diversity. The accurate recognition of alter- OPEN ACCESS native splice sites is regulated by information contained in networks of protein-protein and Citation: Xing Y, Zhao X, Yu T, Liang D, Li J, Wei G, protein-RNA interactions. However, the mechanisms leading to splice site selection are not et al. (2016) MiasDB: A Database of Molecular fully understood. Although numerous databases have been built to describe AS, molecular Interactions Associated with Alternative Splicing of Human Pre-mRNAs. PLoS ONE 11(5): e0155443. interaction databases associated with AS have only recently emerged. In this study, we doi:10.1371/journal.pone.0155443 present a new database, MiasDB, that provides a description of molecular interactions Editor: Ruben Artero, University of Valencia, SPAIN associated with human AS events. This database covers 938 interactions between human splicing factors, RNA elements, transcription factors, kinases and modified histones for 173 Received: November 19, 2015 human AS events. Every entry includes the interaction partners, interaction type, experi- Accepted: April 28, 2016 mental methods, AS type, tissue specificity or disease-relevant information, a simple Published: May 11, 2016 description of the functionally tested interaction in the AS event and references. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Epigenome-Wide Exploratory Study of Monozygotic Twins Suggests Differentially Methylated Regions to Associate with Hand Grip Strength
Biogerontology (2019) 20:627–647 https://doi.org/10.1007/s10522-019-09818-1 (0123456789().,-volV)( 0123456789().,-volV) RESEARCH ARTICLE Epigenome-wide exploratory study of monozygotic twins suggests differentially methylated regions to associate with hand grip strength Mette Soerensen . Weilong Li . Birgit Debrabant . Marianne Nygaard . Jonas Mengel-From . Morten Frost . Kaare Christensen . Lene Christiansen . Qihua Tan Received: 15 April 2019 / Accepted: 24 June 2019 / Published online: 28 June 2019 Ó The Author(s) 2019 Abstract Hand grip strength is a measure of mus- significant CpG sites or pathways were found, how- cular strength and is used to study age-related loss of ever two of the suggestive top CpG sites were mapped physical capacity. In order to explore the biological to the COL6A1 and CACNA1B genes, known to be mechanisms that influence hand grip strength varia- related to muscular dysfunction. By investigating tion, an epigenome-wide association study (EWAS) of genomic regions using the comb-p algorithm, several hand grip strength in 672 middle-aged and elderly differentially methylated regions in regulatory monozygotic twins (age 55–90 years) was performed, domains were identified as significantly associated to using both individual and twin pair level analyses, the hand grip strength, and pathway analyses of these latter controlling the influence of genetic variation. regions revealed significant pathways related to the Moreover, as measurements of hand grip strength immune system, autoimmune disorders, including performed over 8 years were available in the elderly diabetes type 1 and viral myocarditis, as well as twins (age 73–90 at intake), a longitudinal EWAS was negative regulation of cell differentiation. -

Distinct Genetic Alterations in Colorectal Cancer
Distinct Genetic Alterations in Colorectal Cancer Hassan Ashktorab1*, Alejandro A. Scha¨ffer2, Mohammad Daremipouran3, Duane T. Smoot3, Edward Lee3, Hassan Brim3 1 Department of Medicine and Cancer Center, Howard University, College of Medicine, Washington, DC, United States of America, 2 National Center for Biotechnology Information, National Institutes of Health (NIH), Department of Health and Human Services (DHHS), Bethesda, Maryland, United States of America, 3 Department of Pathology, Howard University, College of Medicine, Washington, DC, United States of America Abstract Background: Colon cancer (CRC) development often includes chromosomal instability (CIN) leading to amplifications and deletions of large DNA segments. Epidemiological, clinical, and cytogenetic studies showed that there are considerable differences between CRC tumors from African Americans (AAs) and Caucasian patients. In this study, we determined genomic copy number aberrations in sporadic CRC tumors from AAs, in order to investigate possible explanations for the observed disparities. Methodology/Principal Findings: We applied genome-wide array comparative genome hybridization (aCGH) using a 105k chip to identify copy number aberrations in samples from 15 AAs. In addition, we did a population comparative analysis with aCGH data in Caucasians as well as with a widely publicized list of colon cancer genes (CAN genes). There was an average of 20 aberrations per patient with more amplifications than deletions. Analysis of DNA copy number of frequently altered chromosomes revealed that deletions occurred primarily in chromosomes 4, 8 and 18. Chromosomal duplications occurred in more than 50% of cases on chromosomes 7, 8, 13, 20 and X. The CIN profile showed some differences when compared to Caucasian alterations. Conclusions/Significance: Chromosome X amplification in male patients and chromosomes 4, 8 and 18 deletions were prominent aberrations in AAs. -

WO 2019/079361 Al 25 April 2019 (25.04.2019) W 1P O PCT
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization I International Bureau (10) International Publication Number (43) International Publication Date WO 2019/079361 Al 25 April 2019 (25.04.2019) W 1P O PCT (51) International Patent Classification: CA, CH, CL, CN, CO, CR, CU, CZ, DE, DJ, DK, DM, DO, C12Q 1/68 (2018.01) A61P 31/18 (2006.01) DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, HN, C12Q 1/70 (2006.01) HR, HU, ID, IL, IN, IR, IS, JO, JP, KE, KG, KH, KN, KP, KR, KW, KZ, LA, LC, LK, LR, LS, LU, LY, MA, MD, ME, (21) International Application Number: MG, MK, MN, MW, MX, MY, MZ, NA, NG, NI, NO, NZ, PCT/US2018/056167 OM, PA, PE, PG, PH, PL, PT, QA, RO, RS, RU, RW, SA, (22) International Filing Date: SC, SD, SE, SG, SK, SL, SM, ST, SV, SY, TH, TJ, TM, TN, 16 October 2018 (16. 10.2018) TR, TT, TZ, UA, UG, US, UZ, VC, VN, ZA, ZM, ZW. (25) Filing Language: English (84) Designated States (unless otherwise indicated, for every kind of regional protection available): ARIPO (BW, GH, (26) Publication Language: English GM, KE, LR, LS, MW, MZ, NA, RW, SD, SL, ST, SZ, TZ, (30) Priority Data: UG, ZM, ZW), Eurasian (AM, AZ, BY, KG, KZ, RU, TJ, 62/573,025 16 October 2017 (16. 10.2017) US TM), European (AL, AT, BE, BG, CH, CY, CZ, DE, DK, EE, ES, FI, FR, GB, GR, HR, HU, ΓΕ , IS, IT, LT, LU, LV, (71) Applicant: MASSACHUSETTS INSTITUTE OF MC, MK, MT, NL, NO, PL, PT, RO, RS, SE, SI, SK, SM, TECHNOLOGY [US/US]; 77 Massachusetts Avenue, TR), OAPI (BF, BJ, CF, CG, CI, CM, GA, GN, GQ, GW, Cambridge, Massachusetts 02139 (US). -

Proteomic Analysis of HIV-1 Nef Cellular Binding Partners Reveals a Role for Exocyst Complex Proteins in Mediating Enhancement of Intercellular Nanotube Formation
Proteomic analysis of HIV-1 Nef cellular binding partners reveals a role for exocyst complex proteins in mediating enhancement of intercellular nanotube formation The Harvard community has made this article openly available. Please share how this access benefits you. Your story matters Citation Mukerji, Joya, Kevin C Olivieri, Vikas Misra, Kristin A Agopian, and Dana Gabuzda. 2012. Proteomic analysis of hiv-1 nef cellular binding partners reveals a role for exocyst complex proteins in mediating enhancement of intercellular nanotube formation. Retrovirology 9: 33. Published Version doi:10.1186/1742-4690-9-33 Citable link http://nrs.harvard.edu/urn-3:HUL.InstRepos:10445557 Terms of Use This article was downloaded from Harvard University’s DASH repository, and is made available under the terms and conditions applicable to Other Posted Material, as set forth at http:// nrs.harvard.edu/urn-3:HUL.InstRepos:dash.current.terms-of- use#LAA Mukerji et al. Retrovirology 2012, 9:33 http://www.retrovirology.com/content/9/1/33 RESEARCH Open Access Proteomic analysis of HIV-1 Nef cellular binding partners reveals a role for exocyst complex proteins in mediating enhancement of intercellular nanotube formation Joya Mukerji1,2, Kevin C Olivieri1, Vikas Misra1, Kristin A Agopian1,2 and Dana Gabuzda1,2,3* Abstract Background: HIV-1 Nef protein contributes to pathogenesis via multiple functions that include enhancement of viral replication and infectivity, alteration of intracellular trafficking, and modulation of cellular signaling pathways. Nef stimulates formation of tunneling nanotubes and virological synapses, and is transferred to bystander cells via these intercellular contacts and secreted microvesicles. Nef associates with and activates Pak2, a kinase that regulates T-cell signaling and actin cytoskeleton dynamics, but how Nef promotes nanotube formation is unknown. -

The Emerging Role of Ncrnas and RNA-Binding Proteins in Mitotic Apparatus Formation
non-coding RNA Review The Emerging Role of ncRNAs and RNA-Binding Proteins in Mitotic Apparatus Formation Kei K. Ito, Koki Watanabe and Daiju Kitagawa * Department of Physiological Chemistry, Graduate School of Pharmaceutical Science, The University of Tokyo, Bunkyo, Tokyo 113-0033, Japan; [email protected] (K.K.I.); [email protected] (K.W.) * Correspondence: [email protected] Received: 11 November 2019; Accepted: 13 March 2020; Published: 20 March 2020 Abstract: Mounting experimental evidence shows that non-coding RNAs (ncRNAs) serve a wide variety of biological functions. Recent studies suggest that a part of ncRNAs are critically important for supporting the structure of subcellular architectures. Here, we summarize the current literature demonstrating the role of ncRNAs and RNA-binding proteins in regulating the assembly of mitotic apparatus, especially focusing on centrosomes, kinetochores, and mitotic spindles. Keywords: ncRNA; centrosome; kinetochore; mitotic spindle 1. Introduction Non-coding RNAs (ncRNAs) are defined as a class of RNA molecules that are transcribed from genomic DNA, but not translated into proteins. They are mainly classified into the following two categories according to their length—small RNA (<200 nt) and long non-coding RNA (lncRNA) (>200 nt). Small RNAs include traditional RNA molecules, such as transfer RNA (tRNA), small nuclear RNA (snRNA), small nucleolar RNA (snoRNA), PIWI-interacting RNA (piRNA), and micro RNA (miRNA), and they have been studied extensively [1]. Research on lncRNA is behind that on small RNA despite that recent transcriptome analysis has revealed that more than 120,000 lncRNAs are generated from the human genome [2–4]. -

Ancient Genomic Regulatory Blocks Are a Major Source for Gene Deserts in Vertebrates After Whole Genome Duplications
Supplementary Information for: Ancient genomic regulatory blocks are a major source for gene deserts in vertebrates after whole genome duplications María Touceda-Suárez, Elizabeth M. Kita, Rafael D. Acemel, Panos N. Firbas, Marta S. Magri, Silvia Naranjo, Juan J. Tena, Jose Luis Gómez-Skarmeta, Ignacio Maeso, Manuel Irimia Corresponding Authors: Manuel Irimia Centre for Genomic Regulation Dr. Aiguader, 88, 08003 Barcelona, Spain e-mail: [email protected] Phone: +34933160212 Fax: +34933160099 Ignacio Maeso Centro Andaluz de Biología del Desarrollo (CABD-CSIC-UPO) Universidad Pablo de Olavide, Crta. Utrera km.1, 41013 Sevilla, España e-mail: [email protected] Phone: +34954348948 Fax: +34954349376 José Luis Gómez-Skarmeta Centro Andaluz de Biología del Desarrollo (CABD-CSIC-UPO) Universidad Pablo de Olavide, Crta. Utrera km.1, 41013 Sevilla, España e-mail: [email protected] Phone: +34954348948 Fax: +34954349376 1 Supplementary Figures Supplementary Figure S1 - Microsyntenic arrangements of ancient multi-bystander GRBs whose bystanders have become differentially retained next to different trans-dev ohnologs. For each case, the arrangement in a slow-evolving deuterostome (Bla, B. lanceolatum; Sko, S. kowalevskii; Spu, S. purpuratus) is provided on top (blue lines), followed by the GRB arrangements conserved in the human genome. 2 Supplementary Figure S2 - Evolution of the Hey-MrpS28-Hdcc2 GRB and its functional characterization in zebrafish. A) Phylogenetic distribution of the GRB across the studied metazoan species. Only B. floridae, S. kowalevskii, C. teleta and T. adhaerens have conserved both bystander genes: Hddc2 (grey) and MrpS28 (white), linked to the trans-dev gene Hey (black arrows). In vertebrates, each of the Hey paralogs has preserved only one of the bystander genes in a reciprocal manner. -
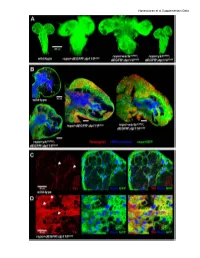
Supplementary Data Vigneswaran Et Al Supplementary Data
Vigneswaran et al Supplementary Data Vigneswaran et al Supplementary Data Figure S1: Yki is required for EGFR-PI3K-driven glial neoplasia in Drosophila (A) Optical projections of whole brain-nerve cord complexes from 3rd instar larvae approximately 130 hrs old. Dorsal view; anterior up. CD8-GFP (green) labels glial cell bodies. Compared to repo>dEGFRλ;dp110CAAX, warts knockdown (repo>wartsdsRNA; dEGFRλ;dp110CAAX) increased neoplastic brain overgrowth and yki knockdown (repo>ykidsRNA;dEGFRλ;dp110CAAX) decreased neoplastic brain overgrowth. (B) 3 µm optical projections of brain hemispheres, age-matched 3rd instar larvae. Frontal sections; anterior up; midline to left. Repo (red) labels glial cell nuclei; CD8-GFP (green) labels glial cell bodies; anti-HRP (blue) counter-stains for neurons and neuropil. (middle) repo>dEGFRλ;dp110CAAX showed increased glial cell numbers (red nuclei) compared to (upper left) wild-type. Compared to repo>dEGFRλ;dp110CAAX, (right) warts knockdown increased neoplastic glial cell numbers (red nuclei), whereas (lower left) yki knockdown reduced neoplastic glial cell numbers (red nuclei). (C, D) Low levels of Yki protein (red) was observed in wild-type central brain glia (white arrows, left panel in C) compared to high levels of cytoplasmic and nuclear Yki protein in dEGFRλ;dp110CAAX neoplastic glia (white arrows, left panel in D); Repo (blue) labels glial cell nuclei; CD8-GFP (green) labels glial cell bodies. Vigneswaran et al Supplementary Data Figure S2: YAP/TAZ expression confined to RTK-amplified tuMor cells and Maintained in patient-derived xenografts (A) On the left, immunohistochemical (IHC) staining in representative normal brain parenchyma in the cortex where YAP expression and TAZ expression was limited to vascular cells and was not detectable in normal neuronal and glial cells. -

Supplementary Table 8. Cpcp PPI Network Details for Significantly Changed Proteins, As Identified in 3.2, Underlying Each of the Five Functional Domains
Supplementary Table 8. cPCP PPI network details for significantly changed proteins, as identified in 3.2, underlying each of the five functional domains. The network nodes represent each significant protein, followed by the list of interactors. Note that identifiers were converted to gene names to facilitate PPI database queries. Functional Domain Node Interactors Development and Park7 Rack1 differentiation Kcnma1 Atp6v1a Ywhae Ywhaz Pgls Hsd3b7 Development and Prdx6 Ncoa3 differentiation Pla2g4a Sufu Ncf2 Gstp1 Grin2b Ywhae Pgls Hsd3b7 Development and Atp1a2 Kcnma1 differentiation Vamp2 Development and Cntn1 Prnp differentiation Ywhaz Clstn1 Dlg4 App Ywhae Ywhab Development and Rac1 Pak1 differentiation Cdc42 Rhoa Dlg4 Ctnnb1 Mapk9 Mapk8 Pik3cb Sod1 Rrad Epb41l2 Nono Ltbp1 Evi5 Rbm39 Aplp2 Smurf2 Grin1 Grin2b Xiap Chn2 Cav1 Cybb Pgls Ywhae Development and Hbb-b1 Atp5b differentiation Hba Kcnma1 Got1 Aldoa Ywhaz Pgls Hsd3b4 Hsd3b7 Ywhae Development and Myh6 Mybpc3 differentiation Prkce Ywhae Development and Amph Capn2 differentiation Ap2a2 Dnm1 Dnm3 Dnm2 Atp6v1a Ywhab Development and Dnm3 Bin1 differentiation Amph Pacsin1 Grb2 Ywhae Bsn Development and Eef2 Ywhaz differentiation Rpgrip1l Atp6v1a Nphp1 Iqcb1 Ezh2 Ywhae Ywhab Pgls Hsd3b7 Hsd3b4 Development and Gnai1 Dlg4 differentiation Development and Gnao1 Dlg4 differentiation Vamp2 App Ywhae Ywhab Development and Psmd3 Rpgrip1l differentiation Psmd4 Hmga2 Development and Thy1 Syp differentiation Atp6v1a App Ywhae Ywhaz Ywhab Hsd3b7 Hsd3b4 Development and Tubb2a Ywhaz differentiation Nphp4 -

SFPQ Rescues F508del-CFTR Expression and Function in Cystic
www.nature.com/scientificreports OPEN SFPQ rescues F508del‑CFTR expression and function in cystic fbrosis bronchial epithelial cells Parameet Kumar1,5, Dharmendra Kumar Soni1,5, Chaitali Sen1, Mads B. Larsen2, Krystyna Mazan‑Mamczarz3, Yulan Piao3, Supriyo De3, Myriam Gorospe3, Raymond A. Frizzell2 & Roopa Biswas1,4* Cystic fbrosis (CF) occurs as a result of mutations in the cystic fbrosis transmembrane conductance regulator (CFTR) gene, which lead to misfolding, trafcking defects, and impaired function of the CFTR protein. Splicing factor proline/glutamine‑rich (SFPQ) is a multifunctional nuclear RNA‑binding protein (RBP) implicated in the regulation of gene expression pathways and intracellular trafcking. Here, we investigated the role of SFPQ in the regulation of the expression and function of F508del‑CFTR in CF lung epithelial cells. We fnd that the expression of SFPQ is reduced in F508del‑CFTR CF epithelial cells compared to WT‑CFTR control cells. Interestingly, the overexpression of SFPQ in CF cells increases the expression as well as rescues the function of F508del‑CFTR. Further, comprehensive transcriptome analyses indicate that SFPQ plays a key role in activating the mutant F508del‑CFTR by modulating several cellular signaling pathways. This is the frst report on the role of SFPQ in the regulation of expression and function of F508del‑CFTR in CF lung disease. Our fndings provide new insights into SFPQ‑mediated molecular mechanisms and point to possible novel epigenetic therapeutic targets for CF and related pulmonary diseases. Cystic fbrosis (CF) is a common life-limiting autosomal recessive genetic disease. Tis disease occurs as a result of mutations in the cystic fbrosis transmembrane conductance regulator (CFTR) gene. -

Centre for Arab Genomic Studies a Division of Sheikh Hamdan Award for Medical Sciences
Centre for Arab Genomic Studies A Division of Sheikh Hamdan Award for Medical Sciences The atalogue for ransmission enetics in rabs C T G A CTGA Database Exocyst Complex Component 4 Alternative Names defects. Individuals were diagnosed with MKS EXOC4 based on the presence of occipital encephalocele as SEC8, S. Cerevisiae, Homolog of well as any combination of liver fibrosis, cleft SEC8 palate, dysplastic kidneys, polydactyly and early KIAA1699 lethality. DNA from both affected and healthy members was obtained and an autozygome guided Record Category mutation analysis of known MKS genes was carried Gene locus out. However, some families did not have mutations in any of the known MKS genes. In such WHO-ICD cases, an exome sequencing was performed. N/A to gene loci Exomes were then searched for compound heterozygous mutations in known MKS genes. Incidence per 100,000 Live Births Failing that, all detected variants were filtered for N/A to gene loci homozygous novel changes within the autozygome. This resulted in the detection of a novel OMIM Number homozygous mutation c.1733A>G (p.Gln578Arg) 608185 in the EXOC4 gene in one of the affected families. This mutation was not found in dbSNP, 1000 Mode of Inheritance genomes or 200 Saudi controls. In-silico analysis N/A to gene loci by PolyPhen predicted this variant to be ‘probably damaging’ while SIFT predicted it to be Gene Map Locus deleterious. The authors noted that EXOC4 had not 7q33 previously been linked to MKS syndrome and that more studies were needed to confirm this Description association. The EXOC4 gene encodes a protein that forms the exocyst complex along with seven other EXOC References proteins.