Nutrition for the Critically Ill Pediatric Patient with Renal Dysfunction 129
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

BSCI440, Spring 2011, Dr. Higgins
Chapter Guides: http://virtual.yosemite.cc.ca.us/rdroual/Course%20Materials/Physiology%20101/Chapte r%20Notes/Fall%202011/chapter_5%20Fall%202011.htm “One time, we had a guy perform who was supposed to be a master of kung fu. Turns out he was a male stripper. Surprised the hell out of us all.” “If you ever get stung by an anemone off the Great Barrier Reef, I want your last thought as you die to be 'damn, Higgins told me about this shit.'” “They knew the dose for this furry meatloaf; some of you know it as a cat.” “Experience and treachery will always overcome youth and exuberance.” *holding up a shoe* “So this is urine, right?” “Isn't that great? That's almost pornographic!” - in reference to a flowchart “The next time your roommate goes to vomit, run in and check their pupils and blood pressure. It’s all for science….the next time you get the stomach flu, think of me.” Lecture 1: Multicellularity & Homeostasis Homeostasis (Chapter 1) Why multicellularity? o surface:volume problem (nutrients), greater specialization, compartmentalization, homeostasis o homeostasis – maintenance of a constant, optimal internal environment Identify 4 basic cell types in mammalian systems o neuron, muscle, epithelial (exocrine vs endocrine), connective o organ systems: endocrine, nervous, musculoskeletal, cardiovascular, respiratory, urinary (osmotic pressure, pH, & ion content), gastrointestinal (absorb & excrete), reproductive, immune, integumentary Understand concept & examples of homeostasis o by isolating internal body fluids by regulating the composition of the fluids surrounding the cells & tissues, organ systems eliminate the need for each cell to respond to changes in the external environment o system sets desired reference level, and makes adjustment via feedback Design closed loop, negative feedback system to control a physical parameter 1 o levels go beyond set point → error signal to integrating center → regulatory mechanisms activate effectors → response → change in regulated variable o stimulus → sensory system → comparator with reference → effector for output o e.g. -

PATHOLOGY of the RENAL SYSTEM”, I Hope You Guys Like It
ﺑﺴﻢ اﷲ اﻟﺮﺣﻤﻦ اﻟﺮﺣﯿﻢ ھﺬه اﻟﻤﺬﻛﺮة ﻋﺒﺎرة ﻋﻦ إﻋﺎدة ﺗﻨﺴﯿﻖ وإﺿﺎﻓﺔ ﻧﻮﺗﺎت وﻣﻮاﺿﯿﻊ ﻟﻤﺬﻛﺮة زﻣﻼﺋﻨﺎ ﻣﻦ اﻟﺪﻓﻌﺔ اﻟﺴﺎﺑﻘﺔ ٤٢٧ اﻷﻋﺰاء.. ﻟﺘﺘﻮاﻓﻖ ﻣﻊ اﻟﻤﻨﮭﺞ اﻟﻤﻘﺮر ﻣﻦ اﻟﻘﺴﻢ ﺣﺮﺻﻨﺎ ﻓﯿﮭﺎ ﻋﻠﻰ إﻋﺎدة ﺻﯿﺎﻏﺔ ﻛﺜﯿﺮ ﻣﻦ اﻟﺠﻤﻞ ﻟﺘﻜﻮن ﺳﮭﻠﺔ اﻟﻔﮭﻢ وﺳﻠﺴﺔ إن ﺷﺎء اﷲ.. وﺿﻔﻨﺎ ﺑﻌﺾ اﻟﻨﻮﺗﺎت اﻟﻤﮭﻤﺔ وأﺿﻔﻨﺎ ﻣﻮاﺿﯿﻊ ﻣﻮﺟﻮدة ﺑﺎﻟـ curriculum ﺗﻌﺪﯾﻞ ٤٢٨ ﻋﻠﻰ اﻟﻤﺬﻛﺮة ﺑﻮاﺳﻄﺔ اﺧﻮاﻧﻜﻢ: ﻓﺎرس اﻟﻌﺒﺪي ﺑﻼل ﻣﺮوة ﻣﺤﻤﺪ اﻟﺼﻮﯾﺎن أﺣﻤﺪ اﻟﺴﯿﺪ ﺣﺴﻦ اﻟﻌﻨﺰي ﻧﺘﻤﻨﻰ ﻣﻨﮭﺎ اﻟﻔﺎﺋﺪة ﻗﺪر اﻟﻤﺴﺘﻄﺎع، وﻻ ﺗﻨﺴﻮﻧﺎ ﻣﻦ دﻋﻮاﺗﻜﻢ ! 2 After hours, or maybe days, of working hard, WE “THE PATHOLOGY TEAM” are proud to present “PATHOLOGY OF THE RENAL SYSTEM”, I hope you guys like it . Plz give us your prayers. Credits: 1st part = written by Assem “ THe AWesOme” KAlAnTAn revised by A.Z.K 2nd part = written by TMA revised by A.Z.K د.ﺧﺎﻟﺪ اﻟﻘﺮﻧﻲ 3rd part = written by Abo Malik revised by 4th part = written by A.Z.K revised by Assem “ THe AWesOme” KAlAnTAn 5th part = written by The Dude revised by TMA figures were provided by A.Z.K Page styling and figure embedding by: If u find any error, or u want to share any idea then plz, feel free to msg me [email protected] 3 Table of Contents Topic page THE NEPHROTIC SYNDROME 4 Minimal Change Disease 5 MEMBRANOUS GLOMERULONEPHRITIS 7 FOCAL SEGMENTAL GLOMERULOSCLEROSIS 9 MEMBRANOPROLIFERATIVE GLOMERULONEPHRITIS 11 DIABETIC NEPHROPATHY (new) 14 NEPHRITIC SYNDROME 18 Acute Post-infectious GN 19 IgA Nephropathy (Berger Disease) 20 Crescentic GN 22 Chronic GN 24 SLE Nephropathy (new) 26 Allograft rejection of the transplanted kidney (new) 27 Urinary Tract OBSTRUCTION, 28 RENAL STONES 23 HYDRONEPHROSIS -

Circulating Extracellular Vesicles As Biomarkers and Drug Delivery Vehicles in Cardiovascular Diseases
biomolecules Review Circulating Extracellular Vesicles As Biomarkers and Drug Delivery Vehicles in Cardiovascular Diseases Renata Caroline Costa de Freitas 1,2 , Rosario Dominguez Crespo Hirata 2 , Mario Hiroyuki Hirata 2 and Elena Aikawa 1,3,4,* 1 Center for Interdisciplinary Cardiovascular Sciences, Division of Cardiovascular Medicine, Brigham and Women’s Hospital, Harvard Medical School, Boston, MA 02115, USA; [email protected] 2 Department of Clinical and Toxicological Analyses, School of Pharmaceutical Sciences, University of Sao Paulo, Sao Paulo 05508-000, Brazil; [email protected] (R.D.C.H.); [email protected] (M.H.H.) 3 Center for Excellence in Vascular Biology, Division of Cardiovascular Medicine, Brigham and Women’s Hospital, Harvard Medical School, Boston, MA 02115, USA 4 Department of Human Pathology, Sechenov First Moscow State Medical University, 119992 Moscow, Russia * Correspondence: [email protected]; Tel.: +1-617-730-7755 Abstract: Extracellular vesicles (EVs) are composed of a lipid bilayer containing transmembrane and soluble proteins. Subtypes of EVs include ectosomes (microparticles/microvesicles), exosomes, and apoptotic bodies that can be released by various tissues into biological fluids. EV cargo can modulate physiological and pathological processes in recipient cells through near- and long-distance intercellular communication. Recent studies have shown that origin, amount, and internal cargos (nucleic acids, proteins, and lipids) of EVs are variable under different pathological conditions, including cardiovascular diseases (CVD). The early detection and management of CVD reduce premature morbidity and mortality. Circulating EVs have attracted great interest as a potential biomarker for diagnostics and follow-up of CVD. This review highlights the role of circulating EVs Citation: de Freitas, R.C.C.; Hirata, as biomarkers for diagnosis, prognosis, and therapeutic follow-up of CVD, and also for drug delivery. -

Preventing Neurological Complications from Dysnatremias in Children
Pediatr Nephrol (2005) 20:1687–1700 DOI 10.1007/s00467-005-1933-6 REVIEW Michael L. Moritz · J. Carlos Ayus Preventing neurological complications from dysnatremias in children Received: 30 November 2004 / Revised: 28 February 2005 / Accepted: 2 March 2005 / Published online: 4 August 2005 IPNA 2005 Abstract Dysnatremias are among the most common ongoing free-water losses or when mild hypernatremia electrolyte abnormalities encountered in hospitalized pa- (Na>145 mE/l) develops. A group at high-risk for neu- tients. In most cases, a dysnatremia results from improper rological damage from hypernatremia in the outpatient fluid management. Dysnatremias can occasionally result setting is that of the breastfed infant. Breastfed infants in death or permanent neurological damage, a tragic must be monitored closely for insufficient lactation and complication that is usually preventable. In this manu- receive lactation support. Judicious use of infant formula script, we discuss the epidemiology, pathogenesis and supplementation may be called for until problems with prevention and treatment of dysnatremias in children. We lactation can be corrected. report on over 50 patients who have suffered death or neurological injury from hospital-acquired hyponatremia. Keywords Hypernatremia · Hyponatremia · Cerebral The main factor contributing to hyponatremic encepha- edema · Myelinolysis · Fluid therapy lopathy in children is the routine use of hypotonic fluids in patients who have an impaired ability to excrete free- water, due to such causes as the postoperative state, Introduction volume depletion and pulmonary and central nervous system diseases. The appropriate use of 0.9% sodium Dysnatremias are a common electrolyte abnormality in chloride in parenteral fluids would likely prevent most children in both the inpatient and outpatient settings. -

Development of a Software to Determine Disturbances in the Acid-Base Balance in Human Blood
NOTA TÉCNICA Vol. 34, No. 2, Agosto 2013, pp. 175-191 Development of a Software to Determine Disturbances in the Acid-Base Balance in Human Blood A. Reyes-Lazalde∗ ABSTRACT ∗∗ M. Reyes-Monreal In intensive care units, one of the most frequent emergencies occurring ∗ M.E. Pérez-Bonilla in patients in critical condition is acid-base imbalance. The ability ∗ R. Reyes-Luna to proficiently manage such patients is achieved through many years of hospital practice. The correct quantification of such imbalances ∗ Laboratorio de Biología allows for the detection of complex alterations. Although this area Interactiva, Escuela de is fundamental for the clinical management of many patients, non- Biología, BUAP. Blvd. specialist doctors rarely receive the appropriate training. In addition, Valsequillo y Av. San Claudio, the learning required to master this area is difficult for doctors due Edificio 112-A, Col. Jardines to the level of mathematics involved. There are two online support de San Manuel. CP. 72570, programs available on the internet for determining blood pH based on Puebla, Pue. the Stewart Model, along with a spreadsheet for the patient’s collected ∗∗ Dirección General de data. Generally, the calculation of hydrogen ion concentration [H+] Innovación Educativa, BUAP. uses a table of equivalences between pH y [H+] for discontinuous values Blvd. Valsequillo y Av. San of pH, with the Davenport diagram used manually. However, none Claudio, Edificio 1AUB, Col. of these programs unite the methods of classic calculus, chemistry Jardines de San Manuel. CP. and physicochemistry. This study develops software for the teaching 72570, Puebla, Pue. and calculation of acid-base imbalance that combines all the relevant methods, such as the Henderson-Hasselbalch equation, the Siggard- Anderson modified excess base equation, the anion gap calculation, the computational implementation of the Davenport diagram, the calculus of [H+] for any value of pH, the calculus for the compensatory process, and the Stewart Model. -

RENAL BIOPSY and GLOMERULONEPHRITIS by J
Postgrad Med J: first published as 10.1136/pgmj.35.409.604 on 1 November 1959. Downloaded from 604 RENAL BIOPSY AND GLOMERULONEPHRITIS By J. H. Ross, M.D., M.R.C.P. Senior Medical Registrar, The Connaught Hospital, and Assistant, The Mledical Unit, The London Hospital Introduction sufficient to establish a diagnosis in a few condi- Safe methods of percutaneous renal biopsy were tions such as amyloid disease and diabetic first described by Perez Ara (1950) and Iversen glomerulosclerosis, but cortex with at least io and Brun (I95I) who used aspiration techniques. glomeruli is necessary for an accurate assessment Many different methods have since been described; of the renal structure; occasionally, up to 40 the simplest is that of Kark and Muehrcke (1954) glomeruli may be present in a single specimen. who use the Franklin modification of the Vim- The revelation of gross structural changes in Silverman needle. the kidneys of patients who can be recognized Brun and Raaschou (1958) have recently re- clinically as having chronic glomerulonephritis is viewed the majority of publications concerned of little value and does not contribute to manage- with renal biopsy and have concluded that the risk ment or prognosis. In contrast, biopsy specimensProtected by copyright. to the patients is slight. They mentioned three from patients in the early stages of glomerulo- fatalities which have been reported but considered nephritis, particularly those who have developed that, in each case, death was probably due to the the nephrotic syndrome insidiously without disease process rather than the investigation. haematuria or renal failure or who showy no Retroperitoneal haematomata have been recorded evidence of disease other than proteinuria, may in patients with hypertension or uraemia but are provide useful information. -

Study on Acid-Base Balance Disorders and the Relationship
ArchiveNephro-Urol of Mon SID. 2020 May; 12(2):e103567. doi: 10.5812/numonthly.103567. Published online 2020 May 23. Research Article Study on Acid-Base Balance Disorders and the Relationship Between Its Parameters and Creatinine Clearance in Patients with Chronic Renal Failure Tran Pham Van 1, *, Thang Le Viet 2, Minh Hoang Thi 1, Lan Dam Thi Phuong 1, Hang Ho Thi 1, Binh Pham Thai 3, Giang Nguyen Thi Quynh 3, Diep Nong Van 4, Sang Vuong Dai 5 and Hop Vu Minh 1 1Biochemistry Department, Military Hospital 103, Hanoi, Vietnam 2Nephrology and Hemodialysis Department, Military Hospital 103, Hanoi, Vietnam 3National Hospital of Endocrinology, Hanoi, Vietnam 4Biochemistry Department, Backan Hospital, Vietnam 5Biochemistry Department, Thanh Nhan Hospital, Hanoi, Vietnam *Corresponding author: Biochemistry Department, Military Hospital 103, Hanoi, Vietnam. Email: [email protected] Received 2020 May 09; Accepted 2020 May 09. Abstract Objectives: We aimed to determine the parameters of acid-base balance in patients with chronic renal failure (CRF) and the rela- tionship between the parameters evaluating acid-base balance and creatinine clearance. Methods: The current cross-sectional study was conducted on 300 patients with CRF (180 males and 120 females). Clinical examina- tion and blood tests by taking an arterial blood sample for blood gas measurement as well as venous blood for biochemical tests to select study participants were performed. Results: Patients with CRF in the metabolic acidosis group accounted for 74%, other types of disorders were less common. The average pH, PCO2, HCO3, tCO2 and BE of the patient group were 7.35 ± 0.09, 34.28 ± 6.92 mmHg, 20.18 ± 6.06 mmol/L, 21.47 ± 6.48 mmHg and -4.72 ± 6.61 mmol/L respectively. -

Clinical Aspect of Salt and Water Balance
Misadventures in salt & water, as well as in acid-base balance Entertaining you is Friedrich C. Luft, Berlin Pflugers Arch 2015 Don’t just “do something” – stand there • 68 year-old woman presents disoriented at 18:00; had undergone tooth extraction that morning and, aside from a life-long mild bleeding tendency, had been quite normal • BP 130/85, pulse regular, respirations 18/min no localizing findings, no edema • Na 118, K 3.6, glucose 8, urea 4 (all mmol/L) • What now? An oil-immersion field showing a normal neutrophil flanked by two giant platelets (Bernard-Soulier syndrome). She had been given desmopressin. In addition, it had been hot so she was advised to “drink lots of water” Serum-Na depends on TBW, Na and K Water in H2 H2O O Na K Serum Na ≈ Naexch + Kexch Total-body H2O Edelman formula Volume Water out Na K Serum Na ≈ Naexch + Kexch total-body H2O Volume Clearance H2O (e) = V 1 - UNa+UK SNa Lots of spheres = little H2O ClH2O(e) neg When UNa+UK >SNa the ClH2O(e) neg and serum Na must fall Few spheres = much H O 2 When UNa+UK < SNa, the Cl (e) pos Cl (e) pos H2O H2O and serum Na must rise Actually, serum Na increased a little faster than we wanted so we infused some free water Had we given 3% saline, serum Na would have increased even faster Iatrogenic SIADH Clin Kidney J 2013;6:96-97 Paradoxal hyponatremia with isotonic electrolyte infusions • 65 year-old woman has meniscus surgery. At that time her Na was 141 mmol/L. -

Davenport Diagram Pathology > Renal Pathologies > Renal Pathologies
Davenport Diagram Pathology > Renal Pathologies > Renal Pathologies DAVENPORT DIAGRAMS: • Davenport diagrams are graphic displays of acid-base states. • They illustrate the dynamic relationships between arterial blood pH, bicarbonate and non-bicarbonate buffers, and the partial pressure of carbon dioxide. • An isopleth represents all possible combinations of bicarbonate and pH values at a given carbon dioxide partial pressure. 4 simple acid-base disorders prior to compensation Graph Features: - The x-axis tracks pH; the healthy homeostatic arterial blood value = 7.4 - Values less than this reflect acidosis; values higher reflect alkalosis. - The y-axis tracks bicarbonate concentration; the healthy homeostatic value 24 millimolar. - Recall that, as bicarbonate concentration increases, pH becomes more alkaline. - Isopleth for a partial pressure of carbon dioxide of 40 mmHg. - A straight line to represent the combination of all non-bicarbonate buffer titration curves. Disorders that cause the blood to become more acidic. • Metabolic acidosis occurs when the reduction in bicarbonate concentration lowers the pH. - Notice that, because this is a non-respiratory disorder, PaCO2 is unaffected. • Respiratory acidosis occurs when the lungs retain excess carbon dioxide, so the partial pressure of carbon dioxide is elevated above normal, which lowers the pH. - Recall that respiratory acidosis produces an elevated bicarbonate concentration, which is reflected in our graph. Disorders that cause the blood to become alkalotic (aka, basic). • Metabolic alkalosis occurs when bicarbonate concentration is elevated. - As in metabolic acidosis, the PaCO2 remains on the 40 mmHg isopleth. 1 / 4 • Respiratory alkalosis occurs when the lungs release too much carbon dioxide - Lowers the PaCO2 and increases pH. Compensatory Mechanisms • The lungs and kidneys respond to acid-base disorders via compensatory mechanisms that bring pH back to normal. -

Guidelines on Urolithiasis
Guidelines on Urolithiasis C. Türk (chair), T. Knoll (vice-chair), A. Petrik, K. Sarica, A. Skolarikos, M. Straub, C. Seitz © European Association of Urology 2014 TABLE OF CONTENTS PAGE 1. METHODOLOGY 7 1.1 Introduction 7 1.2 Data identification 7 1.3 Evidence sources 7 1.4 Level of evidence and grade of recommendation 7 1.5 Publication history 8 1.5.2 Potential conflict of interest statement 8 1.6 References 8 2. CLASSIFICATION OF STONES 9 2.1 Stone size 9 2.2 Stone location 9 2.3 X-ray characteristics 9 2.4 Aetiology of stone formation 9 2.5 Stone composition 9 2.6 Risk groups for stone formation 10 2.7 References 11 3. DIAGNOSIS 12 3.1 Diagnostic imaging 12 3.1.1 Evaluation of patients with acute flank pain 12 3.1.2 Evaluation of patients for whom further treatment of renal stones is planned 13 3.1.3 References 13 3.2 Diagnostics - metabolism-related 14 3.2.1 Basic laboratory analysis - non-emergency urolithiasis patients 15 3.2.2 Analysis of stone composition 15 3.3 References 16 4. TREATMENT OF PATIENTS WITH RENAL COLIC 16 4.1 Renal colic 16 4.1.1 Pain relief 16 4.1.2 Prevention of recurrent renal colic 16 4.1.3 Recommendations for analgesia during renal colic 17 4.1.4 References 17 4.2 Management of sepsis in obstructed kidney 18 4.2.1 Decompression 18 4.2.2 Further measures 18 4.2.3 References 18 5. STONE RELIEF 19 5.1 Observation of ureteral stones 19 5.1.1 Stone-passage rates 19 5.2 Observation of kidney stones 19 5.3 Medical expulsive therapy (MET) 20 5.3.1 Medical agents 20 5.3.2 Factors affecting success of medical expulsive -

Guidelines on Urolithiasis
GUIDELINES ON UROLITHIASIS (Text update April 2010) Ch. Türk (chairman), T. Knoll (vice-chairman), A. Petrik, K. Sarica, C. Seitz, M. Straub, O. Traxer Eur Urol 2001 Oct;40(4):362-71 Eur Urol 2007 Dec;52(6):1610-31 J Urol 2007 Dec;178(6):2418-34 Methodology This update is based on the 2008 version of the Urolithiasis guidelines produced by the former guidelines panel. Literature searches were carried out in Medline and Embase for ran- domised control trials and systematic reviews published between January 1st, 2008, and November 30, 2009, and sig- nificant differences in diagnosis and treatment or in evidence level were considered. Introduction Urinary stone disease continues to occupy an important place in everyday urological practice. The average lifetime risk of stone formation has been reported in the range of 5-10%. Recurrent stone formation is a common problem with all types of stones and therefore an important part of the medical care of patients with stone disease. Classification and Risk Factors Different categories of stone formers can be identified based Urolithiasis 251 on the chemical composition of the stone and the severity of the disease (Table 1). Special attention must be given to those patients who are at high risk of recurrent stone formation (Table 2). Table 1: Categories of stone formers Stone composition Category Non-calcium Infection stones: INF stones Magnesium ammonium phosphate Ammonium urate Uric acid UR Sodium urate Cystine CY Calcium First time stone former, without residual So stones stone or stone fragments. First time stone former, with residual Sres stone or stone fragments. -
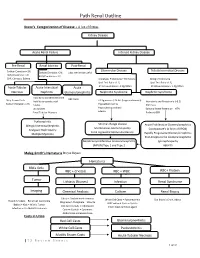
Path Renal Outline
Path Renal Outline Krane’s Categorization of Disease + A lot of Extras Kidney Disease Acute Renal Failure Intrinsic Kidney Disease Pre‐Renal Renal Intrinsic Post‐Renal Sodium Excretion <1% Glomerular Disease Tubulointerstitial Disease Sodium Excretion < 1% Sodium Excretion >2% Labs aren’t that useful BUN/Creatinine > 20 BUN/Creatinine < 10 CHF, Cirrhosis, Edema Urinalysis: Proteinuria + Hematuria Benign Proteinuria Spot Test Ratio >1.5, Spot Test Ratio <1.5, Acute Tubular Acute Interstitial Acute 24 Urine contains > 2.0g/24hrs 24 Urine contains < 1.0g/24hrs Necrosis Nephritis Glomerulonephritis Nephrotic Syndrome Nephritic Syndrome Inability to concentrate Urine RBC Casts Dirty Brown Casts Inability to secrete acid >3.5g protein / 24 hrs (huge proteinuria) Hematuria and Proteinuria (<3.5) Sodium Excretion >2% Edema Hypoalbuminemia RBC Casts Hypercholesterolemia Leukocytes Salt and Water Retention = HTN Focal Tubular Necrosis Edema Reduced GFR Pyelonephritis Minimal change disease Allergic Interstitial Nephritis Acute Proliferative Glomerulonephritis Membranous Glomerulopathy Analgesic Nephropathy Goodpasture’s (a form of RPGN) Focal segmental Glomerulosclerosis Rapidly Progressive Glomerulonephritis Multiple Myeloma Post‐Streptococcal Glomerulonephritis Membranoproliferative Glomerulonephritis IgA nephropathy (MPGN) Type 1 and Type 2 Alport’s Meleg‐Smith’s Hematuria Break Down Hematuria RBCs Only RBC + Crystals RBC + WBC RBC+ Protein Tumor Lithiasis (Stones) Infection Renal Syndrome Imaging Chemical Analysis Culture Renal Biopsy Calcium