International 300 N
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Chapter 3 the Title and Subtitle of This Chapter Convey a Dual Meaning
3.1. Introduction Chapter 3 The title and subtitle of this chapter convey a dual meaning. At first reading, the subtitle Photosynthetic Reaction might seem to indicate that the topic of the structure, function and organization of Centers: photosynthetic reaction centers is So little time, so much to do exceedingly complex and that there is simply insufficient time or space in this brief article to cover the details. While this is John H. Golbeck certainly the case, the subtitle is Department of Biochemistry additionally meant to convey the idea that there is precious little time after the and absorption of a photon to accomplish the Molecular Biology task of preserving the energy in the form of The Pennsylvania State University stable charge separation. University Park, PA 16802 USA The difficulty is there exists a fundamental physical limitation in the amount of time available so that a photochemically induced excited state can be utilized before the energy is invariably wasted. Indeed, the entire design philosophy of biological reaction centers is centered on overcoming this physical, rather than chemical or biological, limitation. In this chapter, I will outline the problem of conserving the free energy of light-induced charge separation by focusing on the following topics: 3.2. Definition of the problem: the need to stabilize a charge-separated state. 3.3. The bacterial reaction center: how the cofactors and proteins cope with this problem in a model system. 3.4. Review of Marcus theory: what governs the rate of electron transfer in proteins? 3.5. Photosystem II: a variation on a theme of the bacterial reaction center. -

Computational Study of Redox Active Centers of Blue Copper Proteins: a Computational DFT Study Matej Pavelka, J.V
Computational Study of Redox Active Centers of Blue Copper Proteins: A Computational DFT Study Matej Pavelka, J.V. Burda To cite this version: Matej Pavelka, J.V. Burda. Computational Study of Redox Active Centers of Blue Copper Proteins: A Computational DFT Study. Molecular Physics, Taylor & Francis, 2009, 106 (24), pp.2733-2748. 10.1080/00268970802672684. hal-00513245 HAL Id: hal-00513245 https://hal.archives-ouvertes.fr/hal-00513245 Submitted on 1 Sep 2010 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Molecular Physics For Peer Review Only Computational Study of Redox Active Centers of Blue Copper Proteins: A Computational DFT Study Journal: Molecular Physics Manuscript ID: TMPH-2008-0332.R1 Manuscript Type: Full Paper Date Submitted by the 07-Dec-2008 Author: Complete List of Authors: Pavelka, Matej; Charles University, Chemical Physics and Optics Burda, J.V.; Charles University, Czech Republic, Department of Chemical physics and optics DFT calculations, plastocyanin, blue copper proteins, copper Keywords: complexes URL: http://mc.manuscriptcentral.com/tandf/tmph Page 1 of 45 Molecular Physics 1 2 3 4 5 Computational Study of Redox Active Centers of Blue Copper 6 7 Proteins: A Computational DFT Study 8 9 10 11 Mat ěj Pavelka and Jaroslav V. -

Glycolysis Citric Acid Cycle Oxidative Phosphorylation Calvin Cycle Light
Stage 3: RuBP regeneration Glycolysis Ribulose 5- Light-Dependent Reaction (Cytosol) phosphate 3 ATP + C6H12O6 + 2 NAD + 2 ADP + 2 Pi 3 ADP + 3 Pi + + 1 GA3P 6 NADP + H Pi NADPH + ADP + Pi ATP 2 C3H4O3 + 2 NADH + 2 H + 2 ATP + 2 H2O 3 CO2 Stage 1: ATP investment ½ glucose + + Glucose 2 H2O 4H + O2 2H Ferredoxin ATP Glyceraldehyde 3- Ribulose 1,5- Light Light Fx iron-sulfur Sakai-Kawada, F Hexokinase phosphate bisphosphate - 4e + center 2016 ADP Calvin Cycle 2H Stroma Mn-Ca cluster + 6 NADP + Light-Independent Reaction Phylloquinone Glucose 6-phosphate + 6 H + 6 Pi Thylakoid Tyr (Stroma) z Fe-S Cyt f Stage 1: carbon membrane Phosphoglucose 6 NADPH P680 P680* PQH fixation 2 Plastocyanin P700 P700* D-(+)-Glucose isomerase Cyt b6 1,3- Pheophytin PQA PQB Fructose 6-phosphate Bisphosphoglycerate ATP Lumen Phosphofructokinase-1 3-Phosphoglycerate ADP Photosystem II P680 2H+ Photosystem I P700 Stage 2: 3-PGA Photosynthesis Fructose 1,6-bisphosphate reduction 2H+ 6 ADP 6 ATP 6 CO2 + 6 H2O C6H12O6 + 6 O2 H+ + 6 Pi Cytochrome b6f Aldolase Plastoquinol-plastocyanin ATP synthase NADH reductase Triose phosphate + + + CO2 + H NAD + CoA-SH isomerase α-Ketoglutarate + Stage 2: 6-carbonTwo 3- NAD+ NADH + H + CO2 Glyceraldehyde 3-phosphate Dihydroxyacetone phosphate carbons Isocitrate α-Ketoglutarate dehydogenase dehydrogenase Glyceraldehyde + Pi + NAD Isocitrate complex 3-phosphate Succinyl CoA Oxidative Phosphorylation dehydrogenase NADH + H+ Electron Transport Chain GDP + Pi 1,3-Bisphosphoglycerate H+ Succinyl CoA GTP + CoA-SH Aconitase synthetase -

Plastocyanin Redox Kinetics in Spinach Chloroplasts: Evidence for Disequilibrium in the High Potential Chain
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Elsevier - Publisher Connector Biochimica et Biophysica Acta 1659 (2004) 63–72 http://www.elsevier.com/locate/bba Plastocyanin redox kinetics in spinach chloroplasts: evidence for disequilibrium in the high potential chain Helmut Kirchhoff*, Mark Aurel Schfttler, Julia Maurer, Engelbert Weis Institut fu¨r Botanik, Schlossgarten 3, D-48149 Mu¨nster, Germany Received 24 May 2004; received in revised form 9 August 2004; accepted 12 August 2004 Available online 26 August 2004 Abstract Reduction kinetics of cytochrome f, plastocyanin (PC) and P700 (dhigh-potential chainT) in thylakoids from spinach were followed after pre-oxidation by a saturating light pulse. We describe a novel approach to follow PC redox kinetics from deconvolution of 810–860 nm absorption changes. The equilibration between the redox-components was analyzed by plotting the redox state of cytochrome f and PC against that of P700. In thylakoids with (1) diminished electron transport rate (adjusted with a cytochrome bf inhibitor) or (2) de-stacked grana, cytochrome f and PC relaxed close to their thermodynamic equilibriums with P700. In stacked thylakoids with non-inhibited electron transport, the equilibration plots were complex and non-hyperbolic, suggesting that during fast electron flux, the dhigh-potential chainT does not homogeneously equilibrate throughout the membrane. Apparent equilibrium constants b5 were calculated, which are below the thermodynamic equilibrium known for the dhigh potential chainT. The disequilibrium found in stacked thylakoids with high electron fluxes is explained by restricted long-range PC diffusion. We develop a model assuming that about 30% of Photosystem I mainly located in grana end- membranes and margins rapidly equilibrate with cytochrome f via short-distance transluminal PC diffusion, while long-range lateral PC migration between grana cores and distant stroma lamellae is restricted. -

Single-Molecule Study of Redox Control Involved in Establishing the Spinach Plastocyanin-Cytochrome B6f Electron Transfer Complex
This is a repository copy of Single-molecule study of redox control involved in establishing the spinach plastocyanin-cytochrome b6f electron transfer complex. White Rose Research Online URL for this paper: http://eprints.whiterose.ac.uk/151341/ Version: Accepted Version Article: Mayneord, G.E., Vasilev, C. orcid.org/0000-0002-0536-882X, Malone, L.A. orcid.org/0000-0002-7625-7545 et al. (3 more authors) (2019) Single-molecule study of redox control involved in establishing the spinach plastocyanin-cytochrome b6f electron transfer complex. Biochimica et Biophysica Acta (BBA) - Bioenergetics, 1860 (7). pp. 591-599. ISSN 0005-2728 https://doi.org/10.1016/j.bbabio.2019.06.013 Article available under the terms of the CC-BY-NC-ND licence (https://creativecommons.org/licenses/by-nc-nd/4.0/). Reuse This article is distributed under the terms of the Creative Commons Attribution-NonCommercial-NoDerivs (CC BY-NC-ND) licence. This licence only allows you to download this work and share it with others as long as you credit the authors, but you can’t change the article in any way or use it commercially. More information and the full terms of the licence here: https://creativecommons.org/licenses/ Takedown If you consider content in White Rose Research Online to be in breach of UK law, please notify us by emailing [email protected] including the URL of the record and the reason for the withdrawal request. [email protected] https://eprints.whiterose.ac.uk/ 1 Single-molecule study of redox control involved in establishing the 2 spinach plastocyanin-cytochrome b6f electron transfer complex 3 Guy E. -

Regulation of Photosynthetic Electron Transport☆
Biochimica et Biophysica Acta 1807 (2011) 375–383 Contents lists available at ScienceDirect Biochimica et Biophysica Acta journal homepage: www.elsevier.com/locate/bbabio Review Regulation of photosynthetic electron transport☆ Jean-David Rochaix ⁎ Department of Molecular Biology, University of Geneva, Geneva, Switzerland Department of Plant Biology, University of Geneva, Geneva, Switzerland article info abstract Article history: The photosynthetic electron transport chain consists of photosystem II, the cytochrome b6 f complex, Received 14 September 2010 photosystem I, and the free electron carriers plastoquinone and plastocyanin. Light-driven charge separation Received in revised form 11 November 2010 events occur at the level of photosystem II and photosystem I, which are associated at one end of the chain Accepted 13 November 2010 with the oxidation of water followed by electron flow along the electron transport chain and concomitant Available online 29 November 2010 pumping of protons into the thylakoid lumen, which is used by the ATP synthase to generate ATP. At the other end of the chain reducing power is generated, which together with ATP is used for CO assimilation. A Keywords: 2 Electron transport remarkable feature of the photosynthetic apparatus is its ability to adapt to changes in environmental Linear electron flow conditions by sensing light quality and quantity, CO2 levels, temperature, and nutrient availability. These Cyclic electron flow acclimation responses involve a complex signaling network in the chloroplasts comprising the thylakoid Photosystem II protein kinases Stt7/STN7 and Stl1/STN7 and the phosphatase PPH1/TAP38, which play important roles in Photosystem I state transitions and in the regulation of electron flow as well as in thylakoid membrane folding. -

Are Plastocyanin and Ferredoxin Specific Electron Carriers Or Generic Redox Capacitors?
1 Are plastocyanin and ferredoxin specific electron carriers or generic redox 2 capacitors? Classical and murburn perspectives on two chloroplast proteins 3 Daniel Andrew Gideon1,2*, Vijay Nirusimhan2 & Kelath Murali Manoj1* 4 *Corresponding authors 5 Email: [email protected]; [email protected] 6 7 1Satyamjayatu: The Science & Ethics Foundation 8 Kulappully, Shoranur-2 (PO), Palakkad District, Kerala State, India-679122. 9 10 2 Department of Biotechnology and Bioinformatics, Bishop Heber College (Autonomous), 11 Vayalur Road, Tiruchirappalli, Tamil Nadu, India-620017. 12 13 ABSTRACT: Within the context of light reaction of photosynthesis, the structure-function 14 correlations of the chloroplast proteins of plastocyanin and ferredoxins (Fd) are analyzed via 15 two perspectives: 1) The Z-scheme, which considers PC/Fd as specific affinity binding-based 16 electron-relay agents, thereby deterministically linking the functions of Cytochrome b6f (Cyt. + 17 b6f) and Photosystem I (PS I) to NADP reduction by Fd:NADPH oxidoreductase (FNR) via 18 protein-protein contacts and 2) The murburn explanation for oxygenic photophosphorylation, 19 which deems PC/Fd as generic ‘redox capacitors’, temporally accepting and releasing one- 20 electron equivalents in reaction milieu. Amino acid residues located on the surface loci of key 21 patches of PC/Fd vary in electrostatic/contour (topography) signatures. Crystal structures of 22 four different complexes each of cyt.f-PC and Fd-FNR show little conservation in the 23 contact-surfaces, thereby discrediting ‘affinity binding-based electron transfers (ET)’ as an 24 evolutionary logic. Further, thermodynamic and kinetic data on wildtype and mutant proteins 25 interactions do not align well with model 1. -

Protein-Protein Interactions and Electron Transfer Associated with Cytochrome F and Plastocyanin from the Cyanobacterium Prochlorothrix Hollandica
PROTEIN-PROTEIN INTERACTIONS AND ELECTRON TRANSFER ASSOCIATED WITH CYTOCHROME F AND PLASTOCYANIN FROM THE CYANOBACTERIUM PROCHLOROTHRIX HOLLANDICA. Maria V. Baranova A Dissertation Submitted to the Graduate College of Bowling Green State University in partial fulfillment of the requirements for the degree of DOCTOR OF PHILOSOPHY August 2007 Committee: George S. Bullerjahn, Advisor J. Devin McAuley Graduate Faculty Representative Michael Y. Ogawa Neocles Leontis ii ABSTRACT George S. Bullerjahn, Advisor This dissertational work describes the minimal structural requirements of interaction surfaces between two proteins involved in photosynthetic electron transfer cyt f and PC from cyanobacterium Prochlorothrix hollandica analyzed by stopped-flow absorption spectroscopy and HSQC NMR. Two mutant P.hollandica cyt f, Y102G and Y102G/F100S, yielding a modified surface-exposed loop region, were expressed and characterized to analyze the structurally unique Prochlorothrix cyt f ‘pocket-like’ region involved in the PC-cyt f complex formation and electron transfer. Stopped-flow studies showed that altering these residues slows down ket more than one order of magnitude. We propose that Tyr102 and Phe100 are actively involved in complex formation between cyt f and PC and serve to minimize distance between electron donor and acceptor. Thus, by removing these residues, the Cu- Fe distance in cyt f -PC complex increases, slowing electron transfer rates. In previous NMR studies(21) it was shown that PC from cyanobacteria interacts with cyt f differently than the comparable proteins in higher plants and algae. The PC-cyt f complex in the cyanobacterium Phormidium laminosum(21) involves a ‘head on’ contact between the hydrophobic (‘northern’) patch of PC with a hydrophobic surface surrounding the cyt f heme, with an average Cu-Fe separation of 15Å. -

Photoreduction of NADP+ by Isolated Reaction Centers of Photosystem II: Requirement for Plastocyanin (Photosynthesis/Electron Transport/Pheophytin) DANIEL 1
Proc. Natl. Acad. Sci. USA Vol. 87, pp. 5930-5934, August 1990 Biophysics Photoreduction of NADP+ by isolated reaction centers of photosystem II: Requirement for plastocyanin (photosynthesis/electron transport/pheophytin) DANIEL 1. ARNON* AND JAMES BARBERt *Department of Plant Biology, University of California, Berkeley, CA 94720; and tAgricultural and Food Research Council Photosynthesis Research Group, Department of Biochemistry, Imperial College, London, United Kingdom Contributed by Daniel I. Arnon, April 30, 1990 ABSTRACT The carrier of photosynthetically generated path and a type in which electrons flow in a linear, noncyclic reducing power is the iron-sulfur protein ferredoxin, which path from water to ferredoxin (2, 4). The light-induced electron provides directly, or via NADP', reducing equivalents needed flow gives rise to electrochemical proton gradients that ac- for CO2 assimilation and other metabolic reactions in the cell. count for the respective "cyclic" and "noncyclic" ATP It is now widely held that, in oxygenic photosynthesis, the formation (13). generation of reduced ferredoxin-NADP+ requires the collab- The specific roles of PSI and PSII in inducing cyclic and oration in series of two photosystems: photosystem II (PSII), noncyclic electron flow are defined by the currently prevalent which energizes electrons to an intermediate reducing potential doctrine embodied in the Z scheme, so called because of the and transfers them to photosystem I (PSI), which in turn is shape of diagrams by which it is represented. The Z scheme solely competent to energize electrons to the strong reducing is widely regarded as the conceptual cornerstone of modern potential required for the reduction offerredoxin-NADP+ (the research in photosynthesis (14). -
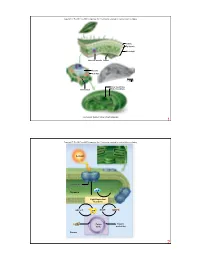
O2 Stroma Photosystem Thylakoid NADP+ ADP + Pi CO2 Sunlight
Copyright © The McGraw-Hill Companies, Inc. Permission required for reproduction or display. Cuticle Epidermis Mesophyll Vascular bundle Stoma Vacuole Cell wall 1.58 µm Inner membrane Outer membrane Chloroplast Courtesy Dr. Kenneth Miller, Brown University 1 Copyright © The McGraw-Hill Companies, Inc. Permission required for reproduction or display. Sunlight Photosystem H2O O2 Thylakoid Light-Dependent Reactions NADP+ NADPH ADP + Pi ATP CO2 Calvin Organic Cycle molecules Stroma 2 Copyright © The McGraw-Hill Companies, Inc. Permission required for reproduction or display. Increasing energy Increasing wavelength 0.001 nm 1 nm 10 nm 1000 nm 0.01 cm 1 cm 1 m 100 m UV Gamma rays X-rays light Infrared Radio waves Visible light 400 nm 430 nm 500 nm 560 nm 600 nm 650 nm 740 nm 3 Copyright © The McGraw-Hill Companies, Inc. Permission required for reproduction or display. high carotenoids chlorophyll a chlorophyll b Light Light Absorbtion Absorbtion low 400 450 500 550 600 650 700 Wavelength (nm) 4 Copyright © The McGraw-Hill Companies, Inc. Permission required for reproduction or display. Chlorophyll a: R = CH R 3 H2C CH H Chlorophyll b: R = CHO H C CH CH 3 2 3 Porphyrin N N head Mg H H N N H3C CH3 H H H O CH2 CO2CH3 CH2 O C O CH2 CH CCH3 CH2 CH2 CH2 CHCH3 Hydrocarbon CH2 tail CH2 CH2 CHCH3 CH2 CH2 CH2 CHCH3 CH3 5 Copyright © The McGraw-Hill Companies, Inc. Permission required for reproduction or display. Oak leaf in summer Oak leaf in autumn © Eric Soder 6 Copyright © The McGraw-Hill Companies, Inc. -

The Structure of the Complex of Plastocyanin And
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Elsevier - Publisher Connector Research Article 323 The structure of the complex of plastocyanin and cytochrome f, determined by paramagnetic NMR and restrained rigid-body molecular dynamics Marcellus Ubbink1†*, Mikael Ejdebäck2, B Göran Karlsson2 and Derek S Bendall1 Background: The reduction of plastocyanin by cytochrome f is part of the Addresses: 1Department of Biochemistry and chain of photosynthetic electron transfer reactions that links photosystems II Cambridge Centre for Molecular Recognition, and I. The reaction is rapid and is influenced by charged residues on both University of Cambridge, Tennis Court Road, Cambridge CB2 1QW, England and 2Department proteins. Previously determined structures show that the plastocyanin copper of Biochemistry and Biophysics, Göteborg and cytochrome f haem redox centres are some distance apart from the University and Chalmers University of Technology, relevant charged sidechains, and until now it was unclear how a transient Lundbergslaboratoriet, Medicinaregatan 9C, S-413 electrostatic complex can be formed that brings the redox centres sufficiently 90, Göteborg, Sweden. close for a rapid reaction. †Present address: Leiden Institute of Chemistry, Leiden University, Gorlaeus Laboratories, P.O. Box Results: A new approach was used to determine the structure of the transient 9502, 2300 RA Leiden, The Netherlands. complex between cytochrome f and plastocyanin. Diamagnetic chemical shift *Corresponding author. changes and intermolecular pseudocontact shifts in the NMR spectrum of E-mail: [email protected] plastocyanin were used as input in restrained rigid-body molecular dynamics calculations. An ensemble of ten structures was obtained, in which the root Key words: chemical shift, dynamic complex, mean square deviation of the plastocyanin position relative to cytochrome f is electron transfer, photosynthesis, pseudocontact shift 1.0 Å. -

Metalloproteins in the Biology of Heterocysts
life Review Metalloproteins in the Biology of Heterocysts Rafael Pernil 1,* and Enrico Schleiff 1,2,3 1 Institute for Molecular Biosciences, Goethe University Frankfurt, Max-von-Laue-Straβe 9, 60438 Frankfurt am Main, Germany; schleiff@bio.uni-frankfurt.de 2 Frankfurt Institute for Advanced Studies, Ruth-Moufang-Straße 1, 60438 Frankfurt am Main, Germany 3 Buchmann Institute for Molecular Life Sciences, Goethe University Frankfurt, Max-von-Laue-Straβe 15, 60438 Frankfurt am Main, Germany * Correspondence: [email protected]; Tel.: +49-69-798-29285 Received: 30 January 2019; Accepted: 28 March 2019; Published: 3 April 2019 Abstract: Cyanobacteria are photoautotrophic microorganisms present in almost all ecologically niches on Earth. They exist as single-cell or filamentous forms and the latter often contain specialized cells for N2 fixation known as heterocysts. Heterocysts arise from photosynthetic active vegetative cells by multiple morphological and physiological rearrangements including the absence of O2 evolution and CO2 fixation. The key function of this cell type is carried out by the metalloprotein complex known as nitrogenase. Additionally, many other important processes in heterocysts also depend on metalloproteins. This leads to a high metal demand exceeding the one of other bacteria in content and concentration during heterocyst development and in mature heterocysts. This review provides an overview on the current knowledge of the transition metals and metalloproteins required by heterocysts in heterocyst-forming cyanobacteria. It discusses the molecular, physiological, and physicochemical properties of metalloproteins involved in N2 fixation, H2 metabolism, electron transport chains, oxidative stress management, storage, energy metabolism, and metabolic networks in the diazotrophic filament. This provides a detailed and comprehensive picture on the heterocyst demands for Fe, Cu, Mo, Ni, Mn, V, and Zn as cofactors for metalloproteins and highlights the importance of such metalloproteins for the biology of cyanobacterial heterocysts.