SARS-Cov2 Enables Anaerobic Bacteria
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Influence of Probiotics on the Firmicutes/Bacteroidetes Ratio In
microorganisms Review The Influence of Probiotics on the Firmicutes/Bacteroidetes Ratio in the Treatment of Obesity and Inflammatory Bowel disease Spase Stojanov 1,2, Aleš Berlec 1,2 and Borut Štrukelj 1,2,* 1 Faculty of Pharmacy, University of Ljubljana, SI-1000 Ljubljana, Slovenia; [email protected] (S.S.); [email protected] (A.B.) 2 Department of Biotechnology, Jožef Stefan Institute, SI-1000 Ljubljana, Slovenia * Correspondence: borut.strukelj@ffa.uni-lj.si Received: 16 September 2020; Accepted: 31 October 2020; Published: 1 November 2020 Abstract: The two most important bacterial phyla in the gastrointestinal tract, Firmicutes and Bacteroidetes, have gained much attention in recent years. The Firmicutes/Bacteroidetes (F/B) ratio is widely accepted to have an important influence in maintaining normal intestinal homeostasis. Increased or decreased F/B ratio is regarded as dysbiosis, whereby the former is usually observed with obesity, and the latter with inflammatory bowel disease (IBD). Probiotics as live microorganisms can confer health benefits to the host when administered in adequate amounts. There is considerable evidence of their nutritional and immunosuppressive properties including reports that elucidate the association of probiotics with the F/B ratio, obesity, and IBD. Orally administered probiotics can contribute to the restoration of dysbiotic microbiota and to the prevention of obesity or IBD. However, as the effects of different probiotics on the F/B ratio differ, selecting the appropriate species or mixture is crucial. The most commonly tested probiotics for modifying the F/B ratio and treating obesity and IBD are from the genus Lactobacillus. In this paper, we review the effects of probiotics on the F/B ratio that lead to weight loss or immunosuppression. -

Ancient Metaproteomics: a Novel Approach for Understanding Disease And
Ancient metaproteomics: a novel approach for understanding disease and diet in the archaeological record Jessica Hendy PhD University of York Archaeology August, 2015 ii Abstract Proteomics is increasingly being applied to archaeological samples following technological developments in mass spectrometry. This thesis explores how these developments may contribute to the characterisation of disease and diet in the archaeological record. This thesis has a three-fold aim; a) to evaluate the potential of shotgun proteomics as a method for characterising ancient disease, b) to develop the metaproteomic analysis of dental calculus as a tool for understanding both ancient oral health and patterns of individual food consumption and c) to apply these methodological developments to understanding individual lifeways of people enslaved during the 19th century transatlantic slave trade. This thesis demonstrates that ancient metaproteomics can be a powerful tool for identifying microorganisms in the archaeological record, characterising the functional profile of ancient proteomes and accessing individual patterns of food consumption with high taxonomic specificity. In particular, analysis of dental calculus may be an extremely valuable tool for understanding the aetiology of past oral diseases. Results of this study highlight the value of revisiting previous studies with more recent methodological approaches and demonstrate that biomolecular preservation can have a significant impact on the effectiveness of ancient proteins as an archaeological tool for this characterisation. Using the approaches developed in this study we have the opportunity to increase the visibility of past diseases and their aetiology, as well as develop a richer understanding of individual lifeways through the production of molecular life histories. iii iv List of Contents Abstract ............................................................................................................................... -

In Vitro Anti-Biofilm and Antibacterial Properties of Streptococcus Downii
microorganisms Article In Vitro Anti-Biofilm and Antibacterial Properties of Streptococcus downii sp. nov. Maigualida Cuenca 1,†, María Carmen Sánchez 1,*,† , Pedro Diz 2, Lucía Martínez-Lamas 3, Maximiliano Álvarez 3, Jacobo Limeres 2 , Mariano Sanz 1 and David Herrera 1 1 ETEP (Etiology and Therapy of Periodontal and Peri-Implant Diseases) Research Group, University Complutense of Madrid (UCM), 28040 Madrid, Spain; [email protected] (M.C.); [email protected] (M.S.); [email protected] (D.H.) 2 Medical-Surgical Dentistry Research Group (OMEQUI), Health Research Institute of Santiago de Compostela (IDIS), University of Santiago de Compostela (USC), 15705 Santiago de Compostela, Spain; [email protected] (P.D.); [email protected] (J.L.) 3 Clinical Microbiology, Microbiology and Infectology Group, Galicia Sur Health Research Institute, Hospital Álvaro Cunqueiro, Complejo Hospitalario Universitario de Vigo, Vigo, 36312 Galicia, Spain; [email protected] (L.M.-L.); [email protected] (M.Á.) * Correspondence: [email protected]; Tel.: +34-913-941-674 † These authors equally contributed to this work. Abstract: The aim of this study was to evaluate the potential anti-biofilm and antibacterial activities of Streptococcus downii sp. nov. To test anti-biofilm properties, Streptococcus mutans, Actinomyces naeslundii, Veillonella parvula, Fusobacterium nucleatum, Porphyromonas gingivalis, and Aggregatibacter actinomycetemcomitans were grown in a biofilm model in the presence or not of S. downii sp. nov. for Citation: Cuenca, M.; Sánchez, M.C.; up to 120 h. For the potential antibacterial activity, 24 h-biofilms were exposed to S. downii sp. nov for Diz, P.; Martínez-Lamas, L.; Álvarez, 24 and 48 h. -

The Oral Microbiome of Healthy Japanese People at the Age of 90
applied sciences Article The Oral Microbiome of Healthy Japanese People at the Age of 90 Yoshiaki Nomura 1,* , Erika Kakuta 2, Noboru Kaneko 3, Kaname Nohno 3, Akihiro Yoshihara 4 and Nobuhiro Hanada 1 1 Department of Translational Research, Tsurumi University School of Dental Medicine, Kanagawa 230-8501, Japan; [email protected] 2 Department of Oral bacteriology, Tsurumi University School of Dental Medicine, Kanagawa 230-8501, Japan; [email protected] 3 Division of Preventive Dentistry, Faculty of Dentistry and Graduate School of Medical and Dental Science, Niigata University, Niigata 951-8514, Japan; [email protected] (N.K.); [email protected] (K.N.) 4 Division of Oral Science for Health Promotion, Faculty of Dentistry and Graduate School of Medical and Dental Science, Niigata University, Niigata 951-8514, Japan; [email protected] * Correspondence: [email protected]; Tel.: +81-45-580-8462 Received: 19 August 2020; Accepted: 15 September 2020; Published: 16 September 2020 Abstract: For a healthy oral cavity, maintaining a healthy microbiome is essential. However, data on healthy microbiomes are not sufficient. To determine the nature of the core microbiome, the oral-microbiome structure was analyzed using pyrosequencing data. Saliva samples were obtained from healthy 90-year-old participants who attended the 20-year follow-up Niigata cohort study. A total of 85 people participated in the health checkups. The study population consisted of 40 male and 45 female participants. Stimulated saliva samples were obtained by chewing paraffin wax for 5 min. The V3–V4 hypervariable regions of the 16S ribosomal RNA (rRNA) gene were amplified by PCR. -

Fatty Acid Diets: Regulation of Gut Microbiota Composition and Obesity and Its Related Metabolic Dysbiosis
International Journal of Molecular Sciences Review Fatty Acid Diets: Regulation of Gut Microbiota Composition and Obesity and Its Related Metabolic Dysbiosis David Johane Machate 1, Priscila Silva Figueiredo 2 , Gabriela Marcelino 2 , Rita de Cássia Avellaneda Guimarães 2,*, Priscila Aiko Hiane 2 , Danielle Bogo 2, Verônica Assalin Zorgetto Pinheiro 2, Lincoln Carlos Silva de Oliveira 3 and Arnildo Pott 1 1 Graduate Program in Biotechnology and Biodiversity in the Central-West Region of Brazil, Federal University of Mato Grosso do Sul, Campo Grande 79079-900, Brazil; [email protected] (D.J.M.); [email protected] (A.P.) 2 Graduate Program in Health and Development in the Central-West Region of Brazil, Federal University of Mato Grosso do Sul, Campo Grande 79079-900, Brazil; pri.fi[email protected] (P.S.F.); [email protected] (G.M.); [email protected] (P.A.H.); [email protected] (D.B.); [email protected] (V.A.Z.P.) 3 Chemistry Institute, Federal University of Mato Grosso do Sul, Campo Grande 79079-900, Brazil; [email protected] * Correspondence: [email protected]; Tel.: +55-67-3345-7416 Received: 9 March 2020; Accepted: 27 March 2020; Published: 8 June 2020 Abstract: Long-term high-fat dietary intake plays a crucial role in the composition of gut microbiota in animal models and human subjects, which affect directly short-chain fatty acid (SCFA) production and host health. This review aims to highlight the interplay of fatty acid (FA) intake and gut microbiota composition and its interaction with hosts in health promotion and obesity prevention and its related metabolic dysbiosis. -

Differentiating Two Closely Related Alexandrium Species Using Comparative Quantitative Proteomics
toxins Article Differentiating Two Closely Related Alexandrium Species Using Comparative Quantitative Proteomics Bryan John J. Subong 1,2,* , Arturo O. Lluisma 1, Rhodora V. Azanza 1 and Lilibeth A. Salvador-Reyes 1,* 1 Marine Science Institute, University of the Philippines- Diliman, Velasquez Street, Quezon City 1101, Philippines; [email protected] (A.O.L.); [email protected] (R.V.A.) 2 Department of Chemistry, The University of Tokyo, 7-3-1 Hongo, Bunkyo City, Tokyo 113-8654, Japan * Correspondence: [email protected] (B.J.J.S.); [email protected] (L.A.S.-R.) Abstract: Alexandrium minutum and Alexandrium tamutum are two closely related harmful algal bloom (HAB)-causing species with different toxicity. Using isobaric tags for relative and absolute quantita- tion (iTRAQ)-based quantitative proteomics and two-dimensional differential gel electrophoresis (2D-DIGE), a comprehensive characterization of the proteomes of A. minutum and A. tamutum was performed to identify the cellular and molecular underpinnings for the dissimilarity between these two species. A total of 1436 proteins and 420 protein spots were identified using iTRAQ-based proteomics and 2D-DIGE, respectively. Both methods revealed little difference (10–12%) between the proteomes of A. minutum and A. tamutum, highlighting that these organisms follow similar cellular and biological processes at the exponential stage. Toxin biosynthetic enzymes were present in both organisms. However, the gonyautoxin-producing A. minutum showed higher levels of osmotic growth proteins, Zn-dependent alcohol dehydrogenase and type-I polyketide synthase compared to the non-toxic A. tamutum. Further, A. tamutum had increased S-adenosylmethionine transferase that may potentially have a negative feedback mechanism to toxin biosynthesis. -

Xiexin Tang Improves the Symptom of Type 2 Diabetic Rats by Modulation of the Gut Microbiota
www.nature.com/scientificreports OPEN Xiexin Tang improves the symptom of type 2 diabetic rats by modulation of the gut microbiota Received: 30 August 2017 Xiaoyan Wei, Jinhua Tao , Suwei Xiao, Shu Jiang, Erxin Shang, Zhenhua Zhu, Dawei Qian Accepted: 13 February 2018 & Jinao Duan Published: xx xx xxxx Type 2 diabetes mellitus (T2DM), a chronic metabolic disease which severely impairs peoples’ quality of life, currently attracted worldwide concerns. There are growing evidences that gut microbiota can exert a great impact on the development of T2DM. Xiexin Tang (XXT), a traditional Chinese medicine prescription, has been clinically used to treat diabetes for thousands of years. However, few researches are investigated on the modulation of gut microbiota community by XXT which will be very helpful to unravel how it works. In this study, bacterial communities were analyzed based on high-throughput 16S rRNA gene sequencing. Results indicated that XXT could notably shape the gut microbiota. T2DM rats treated with XXT exhibited obvious changes in the composition of the gut microbiota, especially for some short chain fatty acids producing and anti-infammatory bacteria such as Adlercreutzia, Alloprevotella, Barnesiella, [Eubacterium] Ventriosum group, Blautia, Lachnospiraceae UCG-001, Papillibacter and Prevotellaceae NK3B31 group. Additionally, XXT could also signifcantly ameliorate hyperglycemia, lipid metabolism dysfunction and infammation in T2DM rats. Moreover, the correlation analysis illustrated that the key microbiota had a close relationship with the T2DM related indexes. The results probably provided useful information for further investigation on its active mechanism and clinical application. T2DM, a chronic metabolic disease characterized by hyperglycemia as a result of insufcient insulin secretion, insulin action or both1, is estimated that its numbers in the adults will increase by 55% by 20352. -

WO 2018/064165 A2 (.Pdf)
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date WO 2018/064165 A2 05 April 2018 (05.04.2018) W !P O PCT (51) International Patent Classification: Published: A61K 35/74 (20 15.0 1) C12N 1/21 (2006 .01) — without international search report and to be republished (21) International Application Number: upon receipt of that report (Rule 48.2(g)) PCT/US2017/053717 — with sequence listing part of description (Rule 5.2(a)) (22) International Filing Date: 27 September 2017 (27.09.2017) (25) Filing Language: English (26) Publication Langi English (30) Priority Data: 62/400,372 27 September 2016 (27.09.2016) US 62/508,885 19 May 2017 (19.05.2017) US 62/557,566 12 September 2017 (12.09.2017) US (71) Applicant: BOARD OF REGENTS, THE UNIVERSI¬ TY OF TEXAS SYSTEM [US/US]; 210 West 7th St., Austin, TX 78701 (US). (72) Inventors: WARGO, Jennifer; 1814 Bissonnet St., Hous ton, TX 77005 (US). GOPALAKRISHNAN, Vanch- eswaran; 7900 Cambridge, Apt. 10-lb, Houston, TX 77054 (US). (74) Agent: BYRD, Marshall, P.; Parker Highlander PLLC, 1120 S. Capital Of Texas Highway, Bldg. One, Suite 200, Austin, TX 78746 (US). (81) Designated States (unless otherwise indicated, for every kind of national protection available): AE, AG, AL, AM, AO, AT, AU, AZ, BA, BB, BG, BH, BN, BR, BW, BY, BZ, CA, CH, CL, CN, CO, CR, CU, CZ, DE, DJ, DK, DM, DO, DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, HN, HR, HU, ID, IL, IN, IR, IS, JO, JP, KE, KG, KH, KN, KP, KR, KW, KZ, LA, LC, LK, LR, LS, LU, LY, MA, MD, ME, MG, MK, MN, MW, MX, MY, MZ, NA, NG, NI, NO, NZ, OM, PA, PE, PG, PH, PL, PT, QA, RO, RS, RU, RW, SA, SC, SD, SE, SG, SK, SL, SM, ST, SV, SY, TH, TJ, TM, TN, TR, TT, TZ, UA, UG, US, UZ, VC, VN, ZA, ZM, ZW. -

Evidence for Persistent and Shared Bacterial Strains Against a Background of Largely Unique Gut Colonization in Hospitalized Premature Infants
The ISME Journal (2016) 10, 2817–2830 © 2016 International Society for Microbial Ecology All rights reserved 1751-7362/16 www.nature.com/ismej ORIGINAL ARTICLE Evidence for persistent and shared bacterial strains against a background of largely unique gut colonization in hospitalized premature infants Tali Raveh-Sadka1, Brian Firek2, Itai Sharon1, Robyn Baker3, Christopher T Brown1, Brian C Thomas1, Michael J Morowitz2 and Jillian F Banfield1 1Department of Earth and Planetary Science, UC Berkeley, Berkeley, CA, USA; 2Department of Surgery, University of Pittsburgh School of Medicine, Pittsburgh, PA, USA and 3Department of Pediatrics, University of Pittsburgh School of Medicine, Pittsburgh, PA, USA The potentially critical stage of initial gut colonization in premature infants occurs in the hospital environment, where infants are exposed to a variety of hospital-associated bacteria. Because few studies of microbial communities are strain-resolved, we know little about the extent to which specific strains persist in the hospital environment and disperse among infants. To study this, we compared 304 near-complete genomes reconstructed from fecal samples of 21 infants hospitalized in the same intensive care unit in two cohorts, over 3 years apart. The genomes represent 159 distinct bacterial strains, only 14 of which occurred in multiple infants. Enterococcus faecalis and Staphylococcus epidermidis, common infant gut colonists, exhibit diversity comparable to that of reference strains, inline with introduction of strains from infant-specific sources rather than a hospital strain pool. Unlike other infants, a pair of sibling infants shared multiple strains, even after extensive antibiotic administration, suggesting overlapping strain-sources and/or genetic selection drive microbiota similarities. -

Gut Microbiome Composition Remains Stable in Individuals with Diabetes-Related Early to Late Stage Chronic Kidney Disease
biomedicines Article Gut Microbiome Composition Remains Stable in Individuals with Diabetes-Related Early to Late Stage Chronic Kidney Disease Ashani Lecamwasam 1,2,3,*, Tiffanie M. Nelson 4, Leni Rivera 3, Elif I. Ekinci 2,5, Richard Saffery 1,6 and Karen M. Dwyer 3 1 Epigenetics Research, Murdoch Children’s Research Institute, VIC 3052, Australia; [email protected] 2 Department of Endocrinology, Austin Health, VIC 3079, Australia; [email protected] 3 School of Medicine, Faculty of Health, Deakin University, VIC 3220, Australia; [email protected] (L.R.); [email protected] (K.M.D.) 4 Menzies Health Institute Queensland, Griffith University, QLD 4222, Australia; [email protected] 5 Department of Medicine, University of Melbourne, VIC 3010, Australia 6 Department of Paediatrics, University of Melbourne, VIC 3010, Australia * Correspondence: [email protected]; Tel.: +613-8341-6200; Fax: +613-9348-1391 Abstract: (1) Background: Individuals with diabetes and chronic kidney disease display gut dysbiosis when compared to healthy controls. However, it is unknown whether there is a change in dysbiosis across the stages of diabetic chronic kidney disease. We investigated a cross-sectional study of patients with early and late diabetes associated chronic kidney disease to identify possible microbial differences between these two groups and across each of the stages of diabetic chronic kidney disease. (2) Methods: This cross-sectional study recruited 95 adults. DNA extracted from collected stool samples were used for 16S rRNA sequencing to identify the bacterial community in the Citation: Lecamwasam, A.; Nelson, gut. (3) Results: The phylum Firmicutes was the most abundant and its mean relative abundance T.M.; Rivera, L.; Ekinci, E.I.; Saffery, was similar in the early and late chronic kidney disease group, 45.99 ± 0.58% and 49.39 ± 0.55%, R.; Dwyer, K.M. -
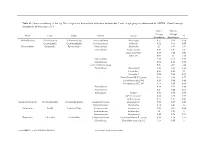
Genus Contributing to the Top 70% of Significant Dissimilarity of Bacteria Between Day 7 and 18 Age Groups As Determined by SIMPER
Table S1: Genus contributing to the top 70% of significant dissimilarity of bacteria between day 7 and 18 age groups as determined by SIMPER. Overall average dissimilarity between ages is 51%. Day 7 Day 18 Average Average Phyla Class Order Family Genus % Abundance Abundance Actinobacteria Actinobacteria Actinomycetales Actinomycetaceae Actinomyces 0.57 0.24 0.74 Coriobacteriia Coriobacteriales Coriobacteriaceae Collinsella 0.51 0.61 0.57 Bacteroidetes Bacteroidia Bacteroidales Bacteroidaceae Bacteroides 2.1 1.63 1.03 Marinifilaceae Butyricimonas 0.82 0.81 0.9 Sanguibacteroides 0.08 0.42 0.59 CAG-873 0.01 1.1 1.68 Marinifilaceae 0.38 0.78 0.91 Marinifilaceae 0.78 1.04 0.97 p-2534-18B5 gut group 0.06 0.7 1.04 Prevotellaceae Alloprevotella 0.46 0.83 0.89 Prevotella 2 0.95 1.11 1.3 Prevotella 7 0.22 0.42 0.56 Prevotellaceae NK3B31 group 0.62 0.68 0.77 Prevotellaceae UCG-003 0.26 0.64 0.89 Prevotellaceae UCG-004 0.11 0.48 0.68 Prevotellaceae 0.64 0.52 0.89 Prevotellaceae 0.27 0.42 0.55 Rikenellaceae Alistipes 0.39 0.62 0.77 dgA-11 gut group 0.04 0.38 0.56 RC9 gut group 0.79 1.17 0.95 Epsilonbacteraeota Campylobacteria Campylobacterales Campylobacteraceae Campylobacter 0.58 0.83 0.97 Helicobacteraceae Helicobacter 0.12 0.41 0.6 Firmicutes Bacilli Lactobacillales Enterococcaceae Enterococcus 0.36 0.31 0.64 Lactobacillaceae Lactobacillus 1.4 1.24 1 Streptococcaceae Streptococcus 0.82 0.58 0.53 Firmicutes Clostridia Clostridiales Christensenellaceae Christensenellaceae R-7 group 0.38 1.36 1.56 Clostridiaceae Clostridium sensu stricto 1 1.16 0.58 -

Longitudinal Characterization of the Gut Bacterial and Fungal Communities in Yaks
Journal of Fungi Article Longitudinal Characterization of the Gut Bacterial and Fungal Communities in Yaks Yaping Wang 1,2,3, Yuhang Fu 3, Yuanyuan He 3, Muhammad Fakhar-e-Alam Kulyar 3 , Mudassar Iqbal 3,4, Kun Li 1,2,* and Jiaguo Liu 1,2,* 1 Institute of Traditional Chinese Veterinary Medicine, College of Veterinary Medicine, Nanjing Agricultural University, Nanjing 210095, China; [email protected] 2 MOE Joint International Research Laboratory of Animal Health and Food Safety, College of Veterinary Medicine, Nanjing Agricultural University, Nanjing 210095, China 3 College of Veterinary Medicine, Huazhong Agricultural University, Wuhan 430070, China; [email protected] (Y.F.); [email protected] (Y.H.); [email protected] (M.F.-e.-A.K.); [email protected] (M.I.) 4 Faculty of Veterinary and Animal Sciences, The Islamia University of Bahawalpur, Bahawalpur 63100, Pakistan * Correspondence: [email protected] (K.L.); [email protected] (J.L.) Abstract: Development phases are important in maturing immune systems, intestinal functions, and metabolism for the construction, structure, and diversity of microbiome in the intestine during the entire life. Characterizing the gut microbiota colonization and succession based on age-dependent effects might be crucial if a microbiota-based therapeutic or disease prevention strategy is adopted. The purpose of this study was to reveal the dynamic distribution of intestinal bacterial and fungal communities across all development stages in yaks. Dynamic changes (a substantial difference) in the structure and composition ratio of the microbial community were observed in yaks that Citation: Wang, Y.; Fu, Y.; He, Y.; matched the natural aging process from juvenile to natural aging.