Diagnosis and Management of Hepatic Encephalopathy
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Encephalopathy and Encephalitis Associated with Cerebrospinal
SYNOPSIS Encephalopathy and Encephalitis Associated with Cerebrospinal Fluid Cytokine Alterations and Coronavirus Disease, Atlanta, Georgia, USA, 2020 Karima Benameur,1 Ankita Agarwal,1 Sara C. Auld, Matthew P. Butters, Andrew S. Webster, Tugba Ozturk, J. Christina Howell, Leda C. Bassit, Alvaro Velasquez, Raymond F. Schinazi, Mark E. Mullins, William T. Hu There are few detailed investigations of neurologic unnecessary staff exposure and difficulties in estab- complications in severe acute respiratory syndrome lishing preillness neurologic status without regular coronavirus 2 infection. We describe 3 patients with family visitors. It is known that neurons and glia ex- laboratory-confirmed coronavirus disease who had en- press the putative SARS-CoV-2 receptor angiotensin cephalopathy and encephalitis develop. Neuroimaging converting enzyme 2 (7), and that the related coro- showed nonenhancing unilateral, bilateral, and midline navirus SARS-CoV (responsible for the 2003 SARS changes not readily attributable to vascular causes. All 3 outbreak) can inoculate the mouse olfactory bulb (8). patients had increased cerebrospinal fluid (CSF) levels If SARS-CoV-2 can enter the central nervous system of anti-S1 IgM. One patient who died also had increased (CNS) directly or through hematogenous spread, ce- levels of anti-envelope protein IgM. CSF analysis also rebrospinal fluid (CSF) changes, including viral RNA, showed markedly increased levels of interleukin (IL)-6, IgM, or cytokine levels, might support CNS infec- IL-8, and IL-10, but severe acute respiratory syndrome coronavirus 2 was not identified in any CSF sample. tion as a cause for neurologic symptoms. We report These changes provide evidence of CSF periinfectious/ clinical, blood, neuroimaging, and CSF findings for postinfectious inflammatory changes during coronavirus 3 patients with laboratory-confirmed COVID-19 and disease with neurologic complications. -

Non-Hepatic Hyperammonaemia: an Important, Potentially Reversible Cause of Encephalopathy
Postgrad Med J 2001;77:717–722 717 Postgrad Med J: first published as 10.1136/pmj.77.913.717 on 1 November 2001. Downloaded from CASE REPORTS Non-hepatic hyperammonaemia: an important, potentially reversible cause of encephalopathy N D Hawkes, G A O Thomas, A Jurewicz, O M Williams, C E M Hillier, I N F McQueen, G Shortland Abstract Case reports The clinical syndrome of encephalopathy CASE 1 is most often encountered in the context of A 20 year old man was admitted to a local hos- decompensated liver disease and the diag- pital with two days of inappropriate behaviour, nosis is usually clear cut. Non-hepatic clumsiness, drowsiness, memory loss, slurred causes of encephalopathy are rarer and speech, and abdominal discomfort. Since the tend to present to a wide range of medical age of 2 years he had suVered recurrent rectal specialties with variable and episodic bleeding and investigation had revealed haem- symptoms. Delay can result in the devel- orrhoids. Bleeding from his rectum had contin- opment of potentially life threatening ued over the years but had been worse recently. complications, such as seizures and coma. On examination he was confused. His Early recognition is vital. A history of Glasgow coma scale score (GCS) was 15/15. similar episodes or clinical risk factors Neurological and general examination was nor- and early assessment of blood ammonia mal, with no stigmata of chronic liver disease. levels help establish the diagnosis. In Investigations showed a leucocytosis (leuco- × 9 addition to adequate supportive care, cyte count 22 10 /l) and serum bilirubin level investigation of the underlying cause of of 32 µmol/l. -

TDP-43 Proteinopathy and Motor Neuron Disease in Chronic Traumatic Encephalopathy
J Neuropathol Exp Neurol Vol. 69, No. 9 Copyright Ó 2010 by the American Association of Neuropathologists, Inc. September 2010 pp. 918Y929 ORIGINAL ARTICLE TDP-43 Proteinopathy and Motor Neuron Disease in Chronic Traumatic Encephalopathy Ann C. McKee, MD, Brandon E. Gavett, PhD, Robert A. Stern, PhD, Christopher J. Nowinski, AB, Robert C. Cantu, MD, Neil W. Kowall, MD, Daniel P. Perl, MD, E. Tessa Hedley-Whyte, MD, Bruce Price, MD, Chris Sullivan, Peter Morin, MD, PhD, Hyo-Soon Lee, MD, Caroline A. Kubilus, Daniel H. Daneshvar, MA, Megan Wulff, MPH, and Andrew E. Budson, MD cord in addition to tau neurofibrillary changes, motor neuron loss, Abstract and corticospinal tract degeneration. The TDP-43 proteinopathy Epidemiological evidence suggests that the incidence of amyo- associated with CTE is similar to that found in frontotemporal lobar trophic lateral sclerosis is increased in association with head injury. degeneration with TDP-43 inclusions, in that widespread regions of Repetitive head injury is also associated with the development of the brain are affected. Akin to frontotemporal lobar degeneration chronic traumatic encephalopathy (CTE), a tauopathy characterized with TDP-43 inclusions, in some individuals with CTE, the TDP-43 by neurofibrillary tangles throughout the brain in the relative absence proteinopathy extends to involve the spinal cord and is associated of A-amyloid deposits. We examined 12 cases of CTE and, in 10, with motor neuron disease. This is the first pathological evidence that found a widespread TAR DNA-binding protein of approximately repetitive head trauma experienced in collision sports might be 43 kd (TDP-43) proteinopathy affecting the frontal and temporal associated with the development of a motor neuron disease. -

Picquestion of the Week:3/09/09
McKechnie Field - Spring Training Home of the Pirates PIC QUESTION OF THE WEEK: 3/09/09 Q: Why is rifaximin used in patients with pouchitis? A: Pouchitis is an inflammation of the internal pouch fashioned from small intestinal tissue during ileal pouch anal anastamosis (IPAA). This surgical procedure bypasses the large intestine and may be employed in patients with ulcerative colitis or Crohn’s disease. A temporary ileostomy is placed to allow the pouch to heal without risk of infection. IPAA is considered preferable to an ostomy for patients suffering from inflammatory bowel diseases refractory to medical treatment. The pouch serves as a collection device for waste, but permits the patient to experience regular bowel movements. It is typical for patients with an internal pouch to experience more frequent (average 6 per day) and watery bowel movements. Pouchitis is the most common complication of IPAA and widely regarded as an idiopathic disease; however, colonization by fecal bacteria may be a contributing factor. The condition occurs most commonly in the first six months following reversal of the ileostomy. Although its frequency decreases after six months, nearly 50% of patients with an IPAA will eventually experience pouchitis. Presenting symptoms include diarrhea, increased frequency of bowel movements, bleeding, abdominal pain, and fever. It can result in dehydration and, in severe cases, hospitalization. Treatment of acute pouchitis generally consists of a two-week course of antibiotic therapy with metronidazole or ciprofloxacin. Approximately 10% of patients do not respond to initial treatment and develop chronic (> 4 weeks) pouchitis. A small number of clinical trials support the potential use of rifaximin for the treatment of refractory or recurrent pouchitis. -

Encephalopathy
Jounal ofNeurology, Neurosurgery, and Psychiatry 1997;62:51-60 51 Operational criteria for the classification of chronic alcoholics: identification of Wemicke's encephalopathy D Caine, G M Halliday, J J Kril, C G Harper Abstract suggest that there can be a complete resolution Objectives-To establish better opera- of the underlying brain abnormality in tional criteria for the diagnosis of Wernicke's encephalopathy, similar to the res- Wernicke's encephalopathy. Current olution of neuropathology in hepatic criteria for diagnosing Wernicke's encephalopathy. However, unlike hepatic encephalopathy require the presence of encephalopathy, in which the clinical signs are three clinical signs (oculomotor abnor- a precursor of the pathology, many patients malities, cerebellar dysfunction, and an with the neuropathology of Wernicke's altered mental state), although it has encephalopathy do not have recorded signs of often been reported that most patients do the classic triad.' 2 This raises the question of not filfil all these criteria. whether the clinical correlates of the patholog- Methods-The clinical histories of 28 ical lesions are being missed during life or alcoholics with neurological and neu- whether the pathology represents clinically ropsychological assessments and defini- silent lesions. A similar situation is seen in tive neuropathological diagnoses were Alzheimer's disease, in which many non- examined to determine clinical signs for demented aged patients show a similar type use in a screening schedule. Operational and distribution of neuropathology to their criteria were then proposed for differenti- demented counterparts. 12 Recently research ating patients with Wernicke's into the pathophysiology of aging and encephalopathy alone or in combination Alzheimer's disease has been successfully with Korsakoff's psychosis or hepatic advanced by the use of operational criteria encephalopathy. -

Primary Biliary Cholangitis: a Brief Overview Justin S
REVIEW Primary Biliary Cholangitis: A Brief Overview Justin S. Louie,* Sirisha Grandhe,* Karen Matsukuma,† and Christopher L. Bowlus* Primary biliary cholangitis (PBC), previously referred to supported by the higher concordance of PBC in monozy- as primary biliary cirrhosis, is the most common chronic gotic compared with dizygotic twins.4 In addition, certain cholestatic autoimmune disease affecting adults in the human leukocyte antigen haplotypes have been associ- United States.1 It is characterized by a hallmark serologic ated with PBC, as well as variants at loci along the inter- signature, antimitochondrial antibody (AMA), and specific leukin-12 (IL-12) immunoregulatory pathway (IL-12A and bile duct pathology with progressive intrahepatic duct de- IL-12RB2 loci).5 struction leading to cholestasis. PBC is potentially fatal and can have both intrahepatic and extrahepatic complications. PATHOGENESIS EPIDEMIOLOGY The primary disease mechanism in PBC is thought to be T cell lymphocyte–mediated injury against intralobu- PBC affects all races and ethnicities; however, it is best lar biliary epithelial cells. This causes progressive destruc- studied in the Caucasian population. The condition pre- tion and eventual disappearance of the intralobular bile dominantly affects women older than 40 years, with a ducts. Molecular mimicry has been proposed as the ini- female/male ratio of 9:1.2 Although the incidence of PBC tiating event in the loss of tolerance primarily to mito- appears to be stable, the overall prevalence of the disease chondrial pyruvate dehydrogenase complex, E2, during is increasing.3 An individual’s genetic susceptibility, epige- which exogenous antigens evoke an immune response netic factors, and certain environmental triggers seem to that recognizes an endogenous (self) antigen inciting an play important roles. -

Diagnosis and Management of Primary Biliary Cholangitis Ticle
REVIEW ArtICLE 1 see related editorial on page x Diagnosis and Management of Primary Biliary Cholangitis TICLE R Zobair M. Younossi, MD, MPH, FACG, AGAF, FAASLD1, David Bernstein, MD, FAASLD, FACG, AGAF, FACP2, Mitchell L. Shifman, MD3, Paul Kwo, MD4, W. Ray Kim, MD5, Kris V. Kowdley, MD6 and Ira M. Jacobson, MD7 Primary biliary cholangitis (PBC) is a chronic, cholestatic, autoimmune disease with a variable progressive course. PBC can cause debilitating symptoms including fatigue and pruritus and, if left untreated, is associated with a high risk of cirrhosis and related complications, liver failure, and death. Recent changes to the PBC landscape include a REVIEW A name change, updated guidelines for diagnosis and treatment as well as new treatment options that have recently become available. Practicing clinicians face many unanswered questions when managing PBC. To assist these healthcare providers in managing patients with PBC, the American College of Gastroenterology (ACG) Institute for Clinical Research & Education, in collaboration with the Chronic Liver Disease Foundation (CLDF), organized a panel of experts to evaluate and summarize the most current and relevant peer-reviewed literature regarding PBC. This, combined with the extensive experience and clinical expertise of this expert panel, led to the formation of this clinical guidance on the diagnosis and management of PBC. Am J Gastroenterol https://doi.org/10.1038/s41395-018-0390-3 INTRODUCTION addition, diagnosis and treatment guidelines are changing and a Primary biliary cholangitis (PBC) is a chronic, cholestatic, auto- number of guidelines have been updated [4, 5]. immune disease with a progressive course that may extend over Because of important changes in the PBC landscape, and a num- many decades. -

Non-Alcoholic Fatty Liver Disease
Non-alcoholic fatty liver disease Description Non-alcoholic fatty liver disease (NAFLD) is a buildup of excessive fat in the liver that can lead to liver damage resembling the damage caused by alcohol abuse, but that occurs in people who do not drink heavily. The liver is a part of the digestive system that helps break down food, store energy, and remove waste products, including toxins. The liver normally contains some fat; an individual is considered to have a fatty liver (hepatic steatosis) if the liver contains more than 5 to 10 percent fat. The fat deposits in the liver associated with NAFLD usually cause no symptoms, although they may cause increased levels of liver enzymes that are detected in routine blood tests. Some affected individuals have abdominal pain or fatigue. During a physical examination, the liver may be found to be slightly enlarged. Between 7 and 30 percent of people with NAFLD develop inflammation of the liver (non- alcoholic steatohepatitis, also known as NASH), leading to liver damage. Minor damage to the liver can be repaired by the body. However, severe or long-term damage can lead to the replacement of normal liver tissue with scar tissue (fibrosis), resulting in irreversible liver disease (cirrhosis) that causes the liver to stop working properly. Signs and symptoms of cirrhosis, which get worse as fibrosis affects more of the liver, include fatigue, weakness, loss of appetite, weight loss, nausea, swelling (edema), and yellowing of the skin and whites of the eyes (jaundice). Scarring in the vein that carries blood into the liver from the other digestive organs (the portal vein) can lead to increased pressure in that blood vessel (portal hypertension), resulting in swollen blood vessels (varices) within the digestive system. -

Vaccinations for Adults with Chronic Liver Disease Or Infection
Vaccinations for Adults with Chronic Liver Disease or Infection This table shows which vaccinations you should have to protect your health if you have chronic hepatitis B or C infection or chronic liver disease (e.g., cirrhosis). Make sure you and your healthcare provider keep your vaccinations up to date. Vaccine Do you need it? Hepatitis A Yes! Your chronic liver disease or infection puts you at risk for serious complications if you get infected with the (HepA) hepatitis A virus. If you’ve never been vaccinated against hepatitis A, you need 2 doses of this vaccine, usually spaced 6–18 months apart. Hepatitis B Yes! If you already have chronic hepatitis B infection, you won’t need hepatitis B vaccine. However, if you have (HepB) hepatitis C or other causes of chronic liver disease, you do need hepatitis B vaccine. The vaccine is given in 2 or 3 doses, depending on the brand. Ask your healthcare provider if you need screening blood tests for hepatitis B. Hib (Haemophilus Maybe. Some adults with certain high-risk conditions, for example, lack of a functioning spleen, need vaccination influenzae type b) with Hib. Talk to your healthcare provider to find out if you need this vaccine. Human Yes! You should get this vaccine if you are age 26 years or younger. Adults age 27 through 45 may also be vacci- papillomavirus nated against HPV after a discussion with their healthcare provider. The vaccine is usually given in 3 doses over a (HPV) 6-month period. Influenza Yes! You need a dose every fall (or winter) for your protection and for the protection of others around you. -

Pharmacologic Treatment of Meningitis and Encephalitis in Adult Patients
Published online: 2019-06-19 THIEME Review Article 145 Pharmacologic Treatment of Meningitis and Encephalitis in Adult Patients Andrew K. Treister1 Ines P. Koerner2 1Department of Neurology, Oregon Health & Science University, Address for correspondence Andrew K. Treister, MD, Department Portland, Oregon, United States of Neurology, Oregon Health & Science University, 3181 SW Sam 2Department of Anesthesiology and Perioperative Medicine, Jackson Park, CR 120, Portland, OR 97239-3098, United States Oregon Health & Science University, Portland, Oregon, (e-mail: [email protected]). United States J Neuroanaesthesiol Crit Care 2019;6:145–152 Abstract Meningitis and encephalitis are two inflammatory, often infectious, disorders of the meninges and the central nervous system. Both are associated with significant morbidity and mortality, and require early and aggressive targeted treatment. This article reviews pharmacologic treatment strategies for infectious meningitis and encephalitis, using Keywords the latest available research and guidelines. It provides an overview of empiric anti- ► meningitis microbial treatment approaches for a variety of organisms, including a discussion of ► encephalitis trends in antibiotic resistance where applicable. Key steps in diagnosis and general ► antibiotic resistance management are briefly reviewed. Introduction care–associated infections are not covered in detail, as they are excellently discussed in a recent guideline statement.4 The terms meningitis and encephalitis comprise a broad array of infectious and inflammatory processes involving the central nervous system (CNS) that carry significant morbidity Literature Review 1,2 and mortality. Meningitis refers to inflammation of PubMed was searched in January 2019 for articles published primarily the meninges, although it may spread to involve the between January 1, 2009, and December 31, 2018. -

Idiopathic Intracranial Hypertension
IDIOPATHIC INTRACRANIAL HYPERTENSION William L Hills, MD Neuro-ophthalmology Oregon Neurology Associates Affiliated Assistant Professor Ophthalmology and Neurology Casey Eye Institute, OHSU No disclosures CASE - 19 YO WOMAN WITH HEADACHES X 3 MONTHS Headaches frontal PMHx: obesity Worse lying down Meds: takes ibuprofen for headaches Wake from sleep Pulsatile tinnitus x 1 month. Vision blacks out transiently when she bends over or sits down EXAMINATION Vision: 20/20 R eye, 20/25 L eye. Neuro: PERRL, no APD, EOMI, VF full to confrontation. Dilated fundoscopic exam: 360 degree blurring of disc margins in both eyes, absent SVP. Formal visual field testing: Enlargement of the blind spot, generalized constriction both eyes. MRI brain: Lumbar puncture: Posterior flattening of Opening pressure 39 the globes cm H20 Empty sella Normal CSF studies otherwise normal Headache improved after LP IDIOPATHIC INTRACRANIAL HYPERTENSION SYNDROME: Increased intracranial pressure without ventriculomegaly or mass lesion Normal CSF composition NOMENCLATURE Idiopathic intracranial hypertension (IIH) Benign intracranial hypertension Pseudotumor cerebri Intracranial hypertension secondary to… DIAGNOSTIC CRITERIA Original criteria have been updated to reflect new imaging modalities: 1492 Friedman and Jacobsen. Neurology 2002; 59: Symptoms and signs reflect only those of - increased ICP or papilledema 1495 Documented increased ICP during LP in lateral decubitus position Normal CSF composition No evidence of mass, hydrocephalus, structural -
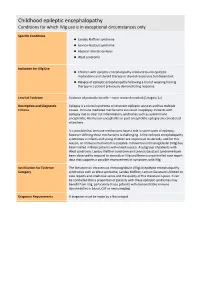
Childhood Epileptic Encephalopathy Conditions for Which Ivig Use Is in Exceptional Circumstances Only
Childhood epileptic encephalopathy Conditions for which IVIg use is in exceptional circumstances only Specific Conditions Landau Kleffner syndrome Lennox‐Gastaut syndrome Atypical rolandic epilepsy West syndrome Indication for IVIg Use Children with epileptic encephalopathy resistant to anti‐epileptic medications and steroid therapy or steroid responsive but dependant Relapse of epileptic encephalopathy following a trial of weaning from Ig therapy in a patient previously demonstrating response Level of Evidence Evidence of probable benefit – more research needed (Category 2a) Description and Diagnostic Epilepsy is a clinical syndrome of recurrent epileptic seizures and has multiple Criteria causes. Immune mediated mechanisms can result in epilepsy. Patients with epilepsy due to clear cut inflammatory syndromes such as autoimmune encephalitis, Rasmussen encephalitis or post encephalitic epilepsy are considered elsewhere. It is possible that immune mechanisms have a role in some cases of epilepsy, however defining these mechanisms is challenging. A few epileptic encephalopathy syndromes in infants and young children are responsive to steroids, and for this reason, an immune mechanism is possible. Intravenous immunoglobulin (IVIg) has been trialled in these patients with mixed success. A subgroup of patients with West syndrome, Landau Kleffner syndrome and Lennox Gaustaut syndrome have been observed to respond to steroids or IVIg and there is uncontrolled case report data that supports a possible improvement of symptoms with IVIg. Justification for Evidence The literature on intravenous immunoglobulin (IVIg) in epileptic encephalopathy Category syndromes such as West syndrome, Landau Kleffner, Lennox Gaustaut is limited to case reports and small case series and the quality of this literature is poor. It can be concluded that a proportion of patients with these epileptic syndromes may benefit from IVIg, particularly those patients with demonstrable immune abnormalities in blood, CSF or neuroimaging.