Detailed Kinetic Study of the Ring Opening of Cycloalkanes by CBS-QB3 Calculations
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Conformations of Cycloalkanes
The Conformations of Cycloalkanes Ring-containing structures are a common occurrence in organic chemistry. We must, therefore spend some time studying the special characteristics of the parent cycloalkanes. Cyclical connectivity imposes constraints on the range of motion that the atoms in rings can undergo. Cyclic molecules are thus more rigid than linear or branched alkanes because cyclic structures have fewer internal degrees of freedom (that is, the motion of one atom greatly influences the motion of the others when they are connected in a ring). In this lesson we will examine structures of the common ring structures found in organic chemistry. The first four cycloalkanes are shown below. cyclopropane cyclobutane cyclopentane cyclohexane The amount of energy stored in a strained ring is estimated by comparing the experimental heat of formation to the calculated heat of formation. The calculated heat of formation is based on the notion that, in the absence of strain, each –CH2– group contributes equally to the heat of formation, in line with the behavior found for the acyclic alkanes (i.e., open chains). Thus, the calculated heat of formation varies linearly with the number of carbon atoms in the ring. Except for the 6-membered ring, the experimental values are found to have a more positive heat of formation than the calculated value owning to ring strain. The plots of calculated and experimental enthalpies of formation and their difference (i.e., ring strain) are seen in the Figure. The 3-membered ring has about 27 kcal/mol of strain. It can be seen that the 6-membered ring possesses almost no ring strain. -

Comparing Models for Measuring Ring Strain of Common Cycloalkanes
The Corinthian Volume 6 Article 4 2004 Comparing Models for Measuring Ring Strain of Common Cycloalkanes Brad A. Hobbs Georgia College Follow this and additional works at: https://kb.gcsu.edu/thecorinthian Part of the Chemistry Commons Recommended Citation Hobbs, Brad A. (2004) "Comparing Models for Measuring Ring Strain of Common Cycloalkanes," The Corinthian: Vol. 6 , Article 4. Available at: https://kb.gcsu.edu/thecorinthian/vol6/iss1/4 This Article is brought to you for free and open access by the Undergraduate Research at Knowledge Box. It has been accepted for inclusion in The Corinthian by an authorized editor of Knowledge Box. Campring Models for Measuring Ring Strain of Common Cycloalkanes Comparing Models for Measuring R..ing Strain of Common Cycloalkanes Brad A. Hobbs Dr. Kenneth C. McGill Chemistry Major Faculty Sponsor Introduction The number of carbon atoms bonded in the ring of a cycloalkane has a large effect on its energy. A molecule's energy has a vast impact on its stability. Determining the most stable form of a molecule is a usefol technique in the world of chemistry. One of the major factors that influ ence the energy (stability) of cycloalkanes is the molecule's ring strain. Ring strain is normally viewed as being directly proportional to the insta bility of a molecule. It is defined as a type of potential energy within the cyclic molecule, and is determined by the level of "strain" between the bonds of cycloalkanes. For example, propane has tl1e highest ring strain of all cycloalkanes. Each of propane's carbon atoms is sp3-hybridized. -

Text Related to Segment 5.02 ©2002 Claude E. Wintner from the Previous Segment We Have the Value of the Heat of Combustion Of
Text Related to Segment 5.02 ©2002 Claude E. Wintner From the previous segment we have the value of the heat of combustion of an "unstrained" methylene unit as -157.4 kcal/mole. On this basis one would predict that combustion of "unstrained" cyclopropane should liberate 3 X 157.4 = 472.2 kcal/mole; however, when cyclopropane is burned, a value of 499.8 kcal/mole is measured for the actual heat of combustion. The difference of 27.6 kcal/mole is interpreted as representing the higher internal energy ("strain energy") of real cyclopropane as compared to a postulated strain-free "model." ("Model" in this usage speaks of a proposal, or postulate.) These facts are graphed conveniently on an energy diagram as follows: "Strain Energy" represents the error units: kcal/mole not to scale in the "strain-free model" real cyclopropane + 4.5 O2 27.6 "strain-free model" of cyclopropane 499.8 observed heat of combustion + 4.5 O2 472.2 3CO2 + 3H2O heat of combustion predicted for "strain-free model" Note that this estimate of the strain energy in cyclopropane amounts to 9.2 kcal/mole of strain per methylene group (dividing 27.6 by 3, because of the three carbon atoms). Comparable values in kcal/mole for other small and medium rings are given in the following table. As one might expect, in general the strain decreases as the ring is enlarged. units: kcal/mole n Total Strain Strain per CH2 3 27.6 9.2 (CH2)n 4 26.3 6.6 5 6.2 1.2 6 0.1 0.0 ! 7 6.2 0.9 8 9.7 1.2 9 12.6 1.4 10 12.4 1.2 12 4.1 0.3 15 1.9 0.1 Without entering into a discussion of the relevant bonding concepts here, and instead relying on geometry alone, interpretation of the source of the strain energy in cyclopropane and cyclobutane is to some extent self-evident. -

Strain-Promoted 1,3-Dipolar Cycloaddition of Cycloalkynes and Organic Azides
Top Curr Chem (Z) (2016) 374:16 DOI 10.1007/s41061-016-0016-4 REVIEW Strain-Promoted 1,3-Dipolar Cycloaddition of Cycloalkynes and Organic Azides 1 1 Jan Dommerholt • Floris P. J. T. Rutjes • Floris L. van Delft2 Received: 24 November 2015 / Accepted: 17 February 2016 / Published online: 22 March 2016 Ó The Author(s) 2016. This article is published with open access at Springerlink.com Abstract A nearly forgotten reaction discovered more than 60 years ago—the cycloaddition of a cyclic alkyne and an organic azide, leading to an aromatic triazole—enjoys a remarkable popularity. Originally discovered out of pure chemical curiosity, and dusted off early this century as an efficient and clean bio- conjugation tool, the usefulness of cyclooctyne–azide cycloaddition is now adopted in a wide range of fields of chemical science and beyond. Its ease of operation, broad solvent compatibility, 100 % atom efficiency, and the high stability of the resulting triazole product, just to name a few aspects, have catapulted this so-called strain-promoted azide–alkyne cycloaddition (SPAAC) right into the top-shelf of the toolbox of chemical biologists, material scientists, biotechnologists, medicinal chemists, and more. In this chapter, a brief historic overview of cycloalkynes is provided first, along with the main synthetic strategies to prepare cycloalkynes and their chemical reactivities. Core aspects of the strain-promoted reaction of cycloalkynes with azides are covered, as well as tools to achieve further reaction acceleration by means of modulation of cycloalkyne structure, nature of azide, and choice of solvent. Keywords Strain-promoted cycloaddition Á Cyclooctyne Á BCN Á DIBAC Á Azide This article is part of the Topical Collection ‘‘Cycloadditions in Bioorthogonal Chemistry’’; edited by Milan Vrabel, Thomas Carell & Floris P. -

Chem 341 • Organic Chemistry I Lecture Summary 10 • September 14, 2007
Chem 341 • Organic Chemistry I Lecture Summary 10 • September 14, 2007 Chapter 4 - Stereochemistry of Alkanes and Cycloalkanes Conformations of Cycloalkanes Cyclic compounds contain something we call Ring Strain. There are three things that contribute to ring strain. Torsional strain (electron repulsion in eclipsing bonds), steric strain (atoms bumping into each other) and angle strain. Angle Strain: the strain due to bond angles being forced to expand or contract from their ideal. Sp3 hybridized atoms want to have bond angles of 109.5°. However, if the rings are very small or very large, there is no way to accommodate this angle. Thus, this increases the energy of the molecule. Heat of Combustion: the amount of heat (energy) released when a molecule burns completely with oxygen. By comparing the heat of combustion of different sized cycloalkanes, their relative energies can be obtained. The fact that the size of the ring has an influence on the total energy of the molecule indicates that there is some degree of instability associated with constraining the rings. This added energy (in addition to what would be expected from carbon and hydrogen combustion per mole) can be attributed to ring strain. n i a r t S g n i R 3 4 5 6 7 8 9 10 11 12 13 14 Ring Size Conformations of Cyclopropane Cyclopropane has a high degree of angle strain due to the highly distorted bond angles. The angle between the carbons is 60°. This cannot be accommodated by sp3-hybridized atoms. Thus, the 60° bonds actually are bent sigma bonds. -

Baeyer's Angle Strain Theory for B.Sc. Sem-II Organic Chemistry: US02CCHE01 by Dr. Vipul B. Kataria Introduction
Baeyer’s Angle Strain Theory For B.Sc. Sem-II Organic Chemistry: US02CCHE01 By Dr. Vipul B. Kataria Introduction Van’t Hoff and Lebel proposed tetrahedral geometry of carbon. The bond angel is of 109˚ 28' (or 109.5˚) for carbon atom in tetrahedral geometry (methane molecule). Baeyer observed different bond angles for different cycloalkanes and also observed some different properties and stability. On this basis, he proposed angle strain theory. The theory explains reactivity and stability of cycloalkanes. Baeyer proposed that the optimum overlap of atomic orbitals is achieved for bond angel of 109.5o. In short, it is ideal bond angle for alkane compounds. Effective and optimum overlap of atomic orbitals produces maximum bond strength and stable molecule. If bond angles deviate from the ideal then ring produce strain. Higher the strain higher the instability. Higher strain produce increased reactivity and increases heat of combustion. Baeyer proposed “any deviation of bond angle from ideal bond angle value (109.5o) will produce a strain in molecule. Higher the deviation lesser the instability”. Que.1 Discuss Baeyer’s angle strain theory using concept of angle strain. Baeyer’s theory is based upon some assumptions as following. 1. All ring systems are planar. Deviation from normal tetrahedral angles results in to instable cycloalkanes. 2. The large ring systems involve negative strain hence do not exists. 3. The bond angles in cyclohexane and higher cycloalkanes (cycloheptane, cyclooctane, cyclononane……..) are not larger than 109.5o because the carbon rings of those compounds are not planar (flat) but they are puckered (Wrinkled). 1 | Page These assumptions are helpful to understand instability of cycloalkane ring systems. -

As a Precursor for Ring Opening and Expansion Dissertation Zur
Peraryl-substituted Cyclotrisilene (c-Si3R4) as a Precursor for Ring Opening and Expansion Dissertation Zur Erlangung des Grades des Doktors der Naturwissenschaften der Naturwissenschaftlich-Technischen Fakultät, der Universität des Saarlandes vorgelegt von M. Sc. Hui Zhao Saarbrücken 08.2018 II Tag des Kolloquiums: 06. 12. 2018 Dekan: Prof. Dr. Guido. Kickelbick Berichterstatter: Prof. Dr. David Scheschkewitz Prof. Dr. Guido Kickelbick Vorsitz: Prof. Dr. Michael Springborg Akademischer Mitarbeiter: Dr. Volker Huch III IV Die vorliegende Arbeit entstand in der Zeit von Oktober 2014 bis August 2018 an der Universität des Saarlandes, Naturwissenschaftlich-Technische Fakultät, Fachrichtung Chemie, im Arbeitskreis von Herrn Prof. Dr. David Scheschkewitz. V VI Kurzzusammenfassung Hauptaugenmerk dieser Arbeit liegt auf dem Untersuchen der Reaktivität von peraryl-substituierten Cyclotrisilenen (c-Si3Tip4) gegenüber verschiedenen kleinen Molekülen. Die Reaktion von c-Si3Tip4 mit Styrol und Diketon liefert zugleich weitere Hinweise auf einen dritten Reaktionsmechanismus, die Ringöffnungsreaktion zu Disilenylsilylenen, die Siliciumversion von vinyl-Carbenen. Die beschriebene Synthese von 1,2,3-Trisilacyclopentadienederivaten, durch die Reaktion von c-Si3Tip4 mit Alkinen, stellt eine neue Methode zur Herstellung von cyclischen, konjugierten C=C−Si=Si Systemen dar. Si3E-bicyclo[1.1.0]butane and Si3E2-bicyclo[1.1.1]pentane (E = S, Se, Te) werden erhalten durch die Additionsreaktionen von c-Si3Tip4 mit Chalkogenen. Die thermische Isomerisierung zu 2-Chalcogena-1,3,4-trisilacyclobutenen wird durch die NMR-spektroskopischen Daten nahegelegt und durch die Isolierung des Hydrolyseprodukts 2-Tellura-1,3,4-trisilacyclobuten gestützt. VII VIII Abstract This thesis concentrates mainly on the reactivity study of a peraryl-substituted cyclotrisilene (c-Si3Tip4) towards various small molecules. -

Energetic Molecules As Future Octane Boosters: Theoretical And
Energetic Molecules as Future Octane Boosters: Theoretical and Experimental Study Dissertation by Mohannad Ali Abdullah Al-Khodaier In Partial Fulfillment of the Requirements For the Degree of Doctor of Philosophy King Abdullah University of Science and Technology Thuwal, Kingdome of Saudi Arabia © June 2018 Mohannad Ali Abdullah Al-Khodaier All Rights Reserved 1 2 EXAMINATION COMMITTEE PAGE The dissertation of Mohannad Ali Abdullah Al-Khodaier is approved by the examination committee. Committee Chairperson: Prof. Mani S. Sarathy Committee Member: Prof. Kazahiro Takanabe, Prof. Luigi Cavallo, Prof. Mohammed Ali Morsy 3 ABSTRACT Energetic Molecules as Future Octane Boosters: Theoretical and Experimental Study Mohannad Ali Abdullah Al-Khodaier The utilization of energetic strained molecules may be one way to mitigate carbon emissions or better and more economical fuel blends. To investigate candidate molecules, limonene and dicyclopentadiene, both theoretical and experimental procedures were implemented here. Computational quantum chemistry methods were employed to determine the thermodynamic properties and kinetic parameters for the hydrogen-abstraction reactions of limonene by a hydrogen atom. Geometry optimization and energy calculation was conducted for all stable species and transition states using Gaussian 09. The rate constants of the H-abstraction reactions were calculated using conventional transition state theory, as implemented in ChemRate software. The obtained values were fitted over the temperature range of 298 – 2000 K to obtain the modified Arrhenius parameters. Increasing the anti-knock quality of gasoline fuels can enable higher efficiency in spark ignition engines. This study explores blending the anti-knock quality of dicyclopentadiene (DCPD, a by-product of ethylene production from naphtha cracking), with various gasoline fuels. -

A Tetra-Amido-Protected Ge5‑Spiropentadiene
Communication Cite This: J. Am. Chem. Soc. 2019, 141, 19252−19256 pubs.acs.org/JACS ‑ A Tetra-amido-Protected Ge5 Spiropentadiene ⊥ † ⊥ † † † ‡ § § Yan Guo, , Zhengqiang Xia, , Jingjing Liu, Jiaxiu Yu, Shenglai Yao, Weiqun Shi, Kongqiu Hu, † † † ‡ † Sanping Chen, Yaoyu Wang, Anyang Li,*, Matthias Driess,*, and Wenyuan Wang*, † Key Laboratory of Synthetic and Natural Functional Molecule Chemistry of Ministry of Education, College of Chemistry and Materials Science, Northwest University, Xi’an, Shaanxi 710069, China ‡ Metalorganics and Inorganic Materials, Department of Chemistry, Technische Universitaẗ Berlin, Straße des 17, Juni 135, Sekr. C2, 10623 Berlin, Germany § Laboratory of Nuclear Energy Chemistry, Institute of High Energy Physics, Chinese Academy of Sciences, Beijing 100049, China *S Supporting Information spiropentadiene B (spiropentasiladiene) was successfully fi ABSTRACT: The rst isolable Ge5-spiropentadiene 1 7 i isolated by Kira and co-workers. Up to now, isolation of was synthesized via the reduction of ( Pr3Si)2NGeCl (3) other E -spiropentadienes (E = Ge, Sn, Pb) has not been with potassium. The crystal structure of 1 reveals a 5 spirocyclic Ge skeleton containing two Ge−Ge double reported. 5 During the past decade, extremely bulky monodentate amido bonds (avg. 2.34 Å), which are fettered in two Ge3 rings with a dihedral angle of 70.193°. The DFT calculations protective groups were more applied in the synthesis of low- and orbital analysis show that the σ-delocalization of the coordinate complexes for main-group elements,8 transition π 9 10 Ge5 skeleton and the 2 -delocalized aromatic Ge3 rings metals, and f-block metals. Herein, we report the synthesis enhance the stability of molecule 1. -
Alicyclic Chemistry (Ehs) Synopsis
ALICYCLIC CHEMISTRY (EHS) SYNOPSIS Textbooks: “Alicyclic Chemistry”, F. J. McQuillin, M. S. Baird, Cambridge, 2nd Ed. 1983. “Alicyclic Chemistry”, M. Grossel, OUP, 1997. “Stereochemistry of Organic Compounds”, E. L. Eliel, S. H. Wilen, Wiley, 1994. “Conformational Analysis”, E. L. Eliel, N. L. Allinger, S. J. Angyal, G. A. Morrison, Interscience, 1967. 1. Ring Strain (a) 1st Lecture: Angle (Baeyer) Strain.- As tetrahedral angle is compressed p- character of ring C-C bond increases. In cyclopropane internal bond 105o with approx. sp3.7 for C-C and sp2.3 for C-H. Thus the C-H shorter and stronger shown by IR, CH acidity, 13C NMR; C-C weaker and longer (π-like) shown by UV, 1H NMR. (b) Torsional (Pitzer) Strain.- Eclipsing of groups along a σ-bond which cannot be relieved by rotation. Planar and puckered cyclobutanes. (c) 2nd Lecture: Transannular Strain.- In medium rings groups project towards one another inside the ring. (d) Cycloalkenes and Cycloalkynes.- Increase in angle strain is balanced to some extent by reduction in torsional strain. Oxirenes, 1H-azirines, 2H-azirines. Trans –cycloalkenes – optical isomerism. 3rd Lecture: Strain measured by Ag+ complexation. Cycloalkyne-Cycloallene equilibrium. 2. Conformational Analysis (Alicyclic only) (a) Thermodynamic Aspects.- Cyclohexane chair and boat. Axial and equatorial hydrogens in chair. Ring flipping equilibrium in monosubstituted cyclohexanes. Rigid trans –decalin and steroid systems (Handout). (b) 4th Lecture: Kinetic Aspects.- (i) Steric control: Base hydrolysis of esters (TS more crowded than SM). Dichromate oxidation of alcohols (TS less crowded than SM). (ii) Stereoelectronic control: E2 elimination. HOBr addition. Epoxide formation (neighbouring group participation). -
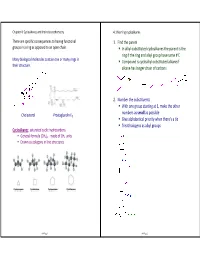
In Alkyl-Substituted Cycloalkanes the Parent Is the Ring If the Ring And
Chapter 4: Cycloalkanes and their stereochemistry 4.1 Naming cycloalkanes There are specific consequences to having functional 1. Find the parent groups in a ring as opposed to an open chain. In alkyl -substituted cycloalkanes the parent is the ring if the ring and alkyl group have same # C Many biological molecules contain one or many rings in Compound is cycloalkyl -substituted alkane if their structure. alkane has longer chain of carbons 2. Number the substituents With one group starting at 1, make the other numbers as small as possible Cholesterol Prostaglandin E 1 Give alphabetical priority when there's a tie Treat halogens as alkyl groups Cycloalkanes : saturated cyclic hydrocarbons General formula (CH 2)n - made of CH 2 units Drawn as polygons in line structures ch4 Page 1 ch4 Page 2 4.2 Cis-trans isomerism in cycloalkanes Stereoisomers sp3 bonds rotate freely in open -chain alkanes, but have Stereoisomers : atoms are connected in same order, but much less rotational freedom in cycloalkanes. differ in three-dimensional orientation recall… Constitutional isomers : same formula but atoms connected differently Cycloalkanes have a "top face" and a "bottom face" when viewed from the side. There are two isomers of 1,2 -dimethylcyclopropane: Name the following molecules, and draw one stereoisomer of each molecule. cis : two groups are pointed towards the same face of a cyclic molecule. trans : two groups are pointed towards opposite faces of a cyclic molecule ch4 Page 3 ch4 Page 4 4.3 Ring strain 4.4 Conformations of cycloalkanes sp 3 carbons have an optimal bond angle of 109.5 o. -

Multi-Configurational Investigation of Thermolytic Pathways of Highly Strained Ring Systems
University of Mississippi eGrove Electronic Theses and Dissertations Graduate School 2012 Multi-Configurational Investigation of Thermolytic Pathways of Highly Strained Ring Systems Jeffrey Dwayne Veals Follow this and additional works at: https://egrove.olemiss.edu/etd Part of the Physical Chemistry Commons Recommended Citation Veals, Jeffrey Dwayne, "Multi-Configurational Investigation of Thermolytic Pathways of Highly Strained Ring Systems" (2012). Electronic Theses and Dissertations. 292. https://egrove.olemiss.edu/etd/292 This Dissertation is brought to you for free and open access by the Graduate School at eGrove. It has been accepted for inclusion in Electronic Theses and Dissertations by an authorized administrator of eGrove. For more information, please contact [email protected]. MULTI-CONFIGURATIONAL INVESTIGATION OF THERMOLYTIC PATHWAYS OF HIGHLY STRAINED RING SYSTEMS JEFFREY D. VEALS A dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy Chemistry University of Mississippi AUGUST 2012 Copyright c AUGUST 2012 by JEFFREY D. VEALS All rights reserved. ABSTRACT The isomerization pathways of model high energy structures are of interest because of their relation to high energy density fuels. Electron resonance has been found to greatly affect the relative activation barriers for several isomerization pathways, and the major goal of this research was to accurately describe its role in determining the relative barriers for strain energy release pathways. This research was centered around the potential energy surfaces (PESs) for σ bond breaking and π bond rotation in these highly strained structures. Of particular interest was how resonance and or electronegativity would affect the allowed or disallowed nature of the activation barriers in these systems.