Ricardaâ•Žs Doktorarbeit
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Atg4b Antibody A
Revision 1 C 0 2 - t Atg4B Antibody a e r o t S Orders: 877-616-CELL (2355) [email protected] Support: 877-678-TECH (8324) 9 9 Web: [email protected] 2 www.cellsignal.com 5 # 3 Trask Lane Danvers Massachusetts 01923 USA For Research Use Only. Not For Use In Diagnostic Procedures. Applications: Reactivity: Sensitivity: MW (kDa): Source: UniProt ID: Entrez-Gene Id: WB H M R Endogenous 48 Rabbit Q9Y4P1 23192 Product Usage Information 2. Ohsumi, Y. (2001) Nat Rev Mol Cell Biol 2, 211-6. 3. Kabeya, Y. et al. (2000) EMBO J 19, 5720-8. Application Dilution 4. Kabeya, Y. et al. (2004) J Cell Sci 117, 2805-12. 5. Mariño, G. et al. (2003) J Biol Chem 278, 3671-8. Western Blotting 1:1000 6. Sou, Y.S. et al. (2008) Mol Biol Cell 19, 4762-75. 7. Hemelaar, J. et al. (2003) J Biol Chem 278, 51841-50. Storage 8. Kabeya, Y. et al. (2004) J Cell Sci 117, 2805-12. 9. Tanida, I. et al. (2004) J Biol Chem 279, 36268-76. Supplied in 10 mM sodium HEPES (pH 7.5), 150 mM NaCl, 100 µg/ml BSA and 50% 10. Fujita, N. et al. (2008) Mol Biol Cell 19, 4651-9. glycerol. Store at –20°C. Do not aliquot the antibody. 11. Fujita, N. et al. (2009) Autophagy 5, 88-9. Specificity / Sensitivity Atg4B Antibody detects endogenous levels of total Atg4B protein. This antibody detects a band at ~27 kDa of unknown origin. Species Reactivity: Human, Mouse, Rat Source / Purification Polyclonal antibodies are produced by immunizing animals with a synthetic peptide corresponding to residues surrounding Ser372 of human Atg4B protein. -

Phosphatidylethanolamine Positively Regulates Autophagy and Longevity
Cell Death and Differentiation (2015) 22, 499–508 OPEN & 2015 Macmillan Publishers Limited All rights reserved 1350-9047/15 www.nature.com/cdd Phosphatidylethanolamine positively regulates autophagy and longevity P Rockenfeller1, M Koska1, F Pietrocola2, N Minois3, O Knittelfelder1, V Sica2, J Franz1, D Carmona-Gutierrez1, G Kroemer*,2,4,5,6,7 and F Madeo*,1,8 Autophagy is a cellular recycling program that retards ageing by efficiently eliminating damaged and potentially harmful organelles and intracellular protein aggregates. Here, we show that the abundance of phosphatidylethanolamine (PE) positively regulates autophagy. Reduction of intracellular PE levels by knocking out either of the two yeast phosphatidylserine decarboxylases (PSD) accelerated chronological ageing-associated production of reactive oxygen species and death. Conversely, the artificial increase of intracellular PE levels, by provision of its precursor ethanolamine or by overexpression of the PE-generating enzyme Psd1, significantly increased autophagic flux, both in yeast and in mammalian cell culture. Importantly administration of ethanolamine was sufficient to extend the lifespan of yeast (Saccharomyces cerevisiae), mammalian cells (U2OS, H4) and flies (Drosophila melanogaster). We thus postulate that the availability of PE may constitute a bottleneck for functional autophagy and that organismal life or healthspan could be positively influenced by the consumption of ethanolamine-rich food. Cell Death and Differentiation (2015) 22, 499–508; doi:10.1038/cdd.2014.219; published online 9 January 2015 Phosphatidylethanolamine (PE) is a phospholipid found in all linked to ageing. Autophagy mainly differs from the proteaso- living organisms. Together with phosphatidylcholine (PC), mal pathway, the other major cellular degradation mechanism, phosphatidylserine (PS) and phosphatidylinositol (PI), PE in two aspects. -

Cysteine Proteases in Protozoan Parasites
UC San Diego UC San Diego Previously Published Works Title Cysteine proteases in protozoan parasites. Permalink https://escholarship.org/uc/item/6sb6c27b Journal PLoS neglected tropical diseases, 12(8) ISSN 1935-2727 Authors Siqueira-Neto, Jair L Debnath, Anjan McCall, Laura-Isobel et al. Publication Date 2018-08-23 DOI 10.1371/journal.pntd.0006512 Peer reviewed eScholarship.org Powered by the California Digital Library University of California REVIEW Cysteine proteases in protozoan parasites Jair L. Siqueira-Neto1*, Anjan Debnath1, Laura-Isobel McCall1¤, Jean A. Bernatchez1, Momar Ndao2,3, Sharon L. Reed4, Philip J. Rosenthal5 1 Center for Discovery and Innovation in Parasitic Diseases, Skaggs School of Pharmacy and Pharmaceutical Sciences, University of California San Diego, La Jolla, California, United States of America, 2 National Reference Centre for Parasitology, The Research Institute of the McGill University Health Center, Montreal, Canada, 3 Program in Infectious Diseases and Immunity in Global Health, The Research Institute of the McGill University Health Centre, Montreal, Quebec, Canada, 4 Departments of Pathology and Medicine, University of California San Diego School of Medicine, La Jolla, California, United States of America, 5 Department of Medicine, University of California, San Francisco, San Francisco, California, a1111111111 United States of America a1111111111 a1111111111 ¤ Current address: Department of Chemistry and Biochemistry, University of Oklahoma, Norman, Oklahoma, a1111111111 United States of America a1111111111 * [email protected] Abstract OPEN ACCESS Cysteine proteases (CPs) play key roles in the pathogenesis of protozoan parasites, includ- Citation: Siqueira-Neto JL, Debnath A, McCall L-I, ing cell/tissue penetration, hydrolysis of host or parasite proteins, autophagy, and evasion Bernatchez JA, Ndao M, Reed SL, et al. -

Serine Proteases with Altered Sensitivity to Activity-Modulating
(19) & (11) EP 2 045 321 A2 (12) EUROPEAN PATENT APPLICATION (43) Date of publication: (51) Int Cl.: 08.04.2009 Bulletin 2009/15 C12N 9/00 (2006.01) C12N 15/00 (2006.01) C12Q 1/37 (2006.01) (21) Application number: 09150549.5 (22) Date of filing: 26.05.2006 (84) Designated Contracting States: • Haupts, Ulrich AT BE BG CH CY CZ DE DK EE ES FI FR GB GR 51519 Odenthal (DE) HU IE IS IT LI LT LU LV MC NL PL PT RO SE SI • Coco, Wayne SK TR 50737 Köln (DE) •Tebbe, Jan (30) Priority: 27.05.2005 EP 05104543 50733 Köln (DE) • Votsmeier, Christian (62) Document number(s) of the earlier application(s) in 50259 Pulheim (DE) accordance with Art. 76 EPC: • Scheidig, Andreas 06763303.2 / 1 883 696 50823 Köln (DE) (71) Applicant: Direvo Biotech AG (74) Representative: von Kreisler Selting Werner 50829 Köln (DE) Patentanwälte P.O. Box 10 22 41 (72) Inventors: 50462 Köln (DE) • Koltermann, André 82057 Icking (DE) Remarks: • Kettling, Ulrich This application was filed on 14-01-2009 as a 81477 München (DE) divisional application to the application mentioned under INID code 62. (54) Serine proteases with altered sensitivity to activity-modulating substances (57) The present invention provides variants of ser- screening of the library in the presence of one or several ine proteases of the S1 class with altered sensitivity to activity-modulating substances, selection of variants with one or more activity-modulating substances. A method altered sensitivity to one or several activity-modulating for the generation of such proteases is disclosed, com- substances and isolation of those polynucleotide se- prising the provision of a protease library encoding poly- quences that encode for the selected variants. -

Proteasy a Jejich Využití V Medicíně Optimalizace Imobilizace Ureasy
UNIVERZITA PALACKÉHO V OLOMOUCI Přírodovědecká fakulta Katedra biochemie Proteasy a jejich využití v medicíně Optimalizace imobilizace ureasy DIPLOMOVÁ PRÁCE Autor: Bc. Jitka Hutařová Studijní program: N1406 Biochemie Studijní obor: Biochemie Forma studia: Prezenční Vedoucí práce: doc. RNDr. Ludmila Zajoncová, Ph.D. Rok: 2016 1 Prohlášení Prohlašuji, že jsem svoji diplomovou práci vypracovala samostatně s využitím informačních zdrojů, které jsou v práci citovány. V Olomouci dne 12. 12. 2016 ……………………………… 2 Poděkování Chtěla bych poděkovat své vedoucí diplomové práce doc. RNDr. Ludmile Zajoncové, Ph.D. za odborné vedení práce, trpělivost a ochotu, kterou mi při zpracování věnovala. Dále bych ráda poděkovala Mgr. Miroslavu Jořenkovi za cenné rady a spolupráci. Poděkováni patří i doc. RNDr. Petru Tarkowskému, Ph.D., který mi umožnil pracovat na plynovém chromatografu. Děkuji také rodině za podporu při studiu. 3 Bibliografická identifikace Jméno a příjmení autora Bc. Jitka Hutařová Název práce Proteasy a jejich využití v medicíně Optimalizace imobilizace ureasy Typ práce Diplomová Pracoviště Katedra biochemie Vedoucí práce doc. RNDr. Ludmila Zajoncová, Ph.D. Rok obhajoby práce 2016 Abstrakt Teoretická část této diplomové práce se zabývá klasifikací proteas, jsou zde popsány jednotlivé rodiny proteas, jejich inhibitory, výskyt a využití, především v medicíně (systémová enzymoterapie). Teoretická část zahrnuje také informace o preparátu Wobenzym, blíže jsou popsány enzymy v něm obsažené (bromelain, papain, trypsin, chymotrypsin). Druhá část je věnována také urease, jejím vlastnostem, výskytu, purifikaci, imobilizaci a praktickým aplikacím. Experimentální část se zabývá optimalizací imobilizace ureasy na magnetické mikročástice Perlosa MG 200 z hlediska volby vhodných podmínek pro imobilizaci. U imobilizované ureasy byla stanovena specifická aktivita, aktivita enzymu během skladování při laboratorní teplotě bez a po ošetření glycinem. -

Diet, Autophagy, and Cancer: a Review
1596 Review Diet, Autophagy, and Cancer: A Review Keith Singletary1 and John Milner2 1Department of Food Science and Human Nutrition, University of Illinois, Urbana, Illinois and 2Nutritional Science Research Group, Division of Cancer Prevention, National Cancer Institute, Bethesda, Maryland Abstract A host of dietary factors can influence various cellular standing of the interactions among bioactive food processes and thereby potentially influence overall constituents, autophagy, and cancer. Whereas a variety cancer risk and tumor behavior. In many cases, these of food components including vitamin D, selenium, factors suppress cancer by stimulating programmed curcumin, resveratrol, and genistein have been shown to cell death. However, death not only can follow the stimulate autophagy vacuolization, it is often difficult to well-characterized type I apoptotic pathway but also can determine if this is a protumorigenic or antitumorigenic proceed by nonapoptotic modes such as type II (macro- response. Additional studies are needed to examine autophagy-related) and type III (necrosis) or combina- dose and duration of exposures and tissue specificity tions thereof. In contrast to apoptosis, the induction of in response to bioactive food components in transgenic macroautophagy may contribute to either the survival or and knockout models to resolve the physiologic impli- death of cells in response to a stressor. This review cations of early changes in the autophagy process. highlights current knowledge and gaps in our under- (Cancer Epidemiol Biomarkers Prev 2008;17(7):1596–610) Introduction A wealth of evidence links diet habits and the accompa- degradation. Paradoxically, depending on the circum- nying nutritional status with cancer risk and tumor stances, this process of ‘‘self-consumption’’ may be behavior (1-3). -

On ATG4B As Drug Target for Treatment of Solid Tumours—The Knowns and the Unknowns
cells Review On ATG4B as Drug Target for Treatment of Solid Tumours—The Knowns and the Unknowns Alexander Agrotis and Robin Ketteler * MRC Laboratory for Molecular Cell Biology, University College London, London WC1E 6BT, UK; [email protected] * Correspondence: [email protected]; Tel.: +44-(0)20-7679-4063 Received: 3 December 2019; Accepted: 19 December 2019; Published: 24 December 2019 Abstract: Autophagy is an evolutionary conserved stress survival pathway that has been shown to play an important role in the initiation, progression, and metastasis of multiple cancers; however, little progress has been made to date in translation of basic research to clinical application. This is partially due to an incomplete understanding of the role of autophagy in the different stages of cancer, and also to an incomplete assessment of potential drug targets in the autophagy pathway. While drug discovery efforts are on-going to target enzymes involved in the initiation phase of the autophagosome, e.g., unc51-like autophagy activating kinase (ULK)1/2, vacuolar protein sorting 34 (Vps34), and autophagy-related (ATG)7, we propose that the cysteine protease ATG4B is a bona fide drug target for the development of anti-cancer treatments. In this review, we highlight some of the recent advances in our understanding of the role of ATG4B in autophagy and its relevance to cancer, and perform a critical evaluation of ATG4B as a druggable cancer target. Keywords: autophagy; ATG4; pancreatic ductal adenocarcinoma (PDAC); drug screening; small molecule compound; screening assay; biomarker 1. Introduction Autophagy is a cellular stress response that has been identified as a target pathway for intervention in various diseases such as neuro-degenerative disorders, pathogen infection, and various types of cancer [1]. -

Huang Y Et Al, PML-Rarα and Autophagy Activation Title Page
Huang Y et al, PML-RARα and autophagy activation Title page PML-RARα enhances autophagic activity through inhibiting Akt/mTOR pathway Running title: PML-RARα and autophagy activation Ying Huang1,2, Xu-Yun Zhao1, Ting-Ting Chen1, Jia-Kai Hou2, Jing Zhang2, Jie Yang1, Scott C. Kogan3 and Guo-Qiang Chen1, 2 Affiliation notes: 1Key Laboratory of Cell Differentiation and Apoptosis of Chinese Ministry of Education, Shanghai Jiao-Tong University School of Medicine (SJTU-SM), Shanghai, China; 2Institute of Health Science, Shanghai Institutes for Biological Sciences (SIBS), Chinese Academy of Sciences & SJTU-SM, Shanghai, China. 3Comprehensive Cancer Center and Department of Laboratory Medicine, University of California, San Francisco, CA. Correspondence: Prof. Guo-Qiang Chen. No.280, Chong-Qing South Rd, Shanghai 200025, CHINA. Tel/Fax: +86-21-64154900; Email: [email protected]. Grant supports: This work is supported in part by grants from National Basic Research Program of China (973 Program) (NO2009CB918404), National Natural Science Foundation of China (NSFC, 30630034, 30870523, 90813034), Chinese Academy of Sciences (KSCX2-YW-R-097), Science and Technology Commission of Shanghai (08JC1413400) as well as leading academic discipline project of Shanghai Municipal Education Commission (J50201). Dr. Guo-Qiang Chen is supported by Shanghai Ling-Jun Talent Program. 1 Huang Y et al, PML-RARα and autophagy activation Abstract Autophagy is a highly conserved, closely-regulated homeostatic cellular activity that allows for the bulk degradation of long-lived proteins and cytoplasmic organelles. Its roles in cancer initiation and progression and in determining the response of tumor cells to anticancer therapy are complicated, and the potential significance of autophagy in the pathogenesis and therapeutic response of acute myeloid leukemia has a little investigation. -

Appendix 1 – Protocol for Preventing Or Reversing Chronic Disease The
Appendix 1 – Protocol for Preventing or Reversing Chronic Disease The first author has developed a protocol over the past decade for preventing or reversing chronic disease based on the following systemic medical principle: “at the present time, removal of cause is a necessary, but not necessarily sufficient, condition for restorative treatment to be effective”[1]. The protocol methodology refines the age-old principles of both reducing harm in addition to providing treatment, and allows better identification of factors that contribute to the disease process (so that they may be eliminated if possible). These contributing factors are expansive and may include a combination of Lifestyle choices (diet, exercise, smoking), iatrogenic and biotoxin exposures, environmental/occupational exposures, and psychosocial stressors. This strategy exploits the existing literature to identify patterns of biologic response using biomarkers from various modalities of diagnostic testing to capture a much broader list of potential contributing factors. Existing inflammatory bowel diseases (IBD) Biomarker Identification to Remove Contributing Factors and Implement Treatment The initial protocol steps are diagnostic. The main output of these diagnostics will be identification of the biomarker levels and symptoms that reflect abnormalities, and the directions of change required to eliminate these abnormalities. In the present study, hundreds of general and specific biomarkers and symptoms were identified from the core IBD literature. The highest frequency (based on numbers of record appearances) biomarkers and symptoms were extracted, and are listed in Table 1 (highest frequency items first, reading down each column before proceeding to the next column). They are not necessarily consensus biomarkers. They are biomarkers whose values were altered by a treatment or contributing factor, and reported in the core IBD literature. -

The Functional and Pathologic Relevance of Autophagy Proteases
The functional and pathologic relevance of autophagy proteases Álvaro F. Fernández, Carlos López-Otín J Clin Invest. 2015;125(1):33-41. https://doi.org/10.1172/JCI73940. Review Autophagy is a well-conserved catabolic process essential for cellular homeostasis. First described in yeast as an adaptive response to starvation, this pathway is also present in higher eukaryotes, where it is triggered by stress signals such as damaged organelles or pathogen infection. Autophagy is characterized at the cellular level by the engulfment of portions of the cytoplasm in double-membrane structures called autophagosomes. Autophagosomes fuse with lysosomes, resulting in degradation of the inner autophagosomal membrane and luminal content. This process is coordinated by complex molecular systems, including the ATG8 ubiquitin–like conjugation system and the ATG4 cysteine proteases, which are implicated in the formation, elongation, and fusion of these autophagic vesicles. In this Review, we focus on the diverse functional roles of the autophagins, a protease family formed by the four mammalian orthologs of yeast Atg4. We also address the dysfunctional expression of these proteases in several pathologic conditions such as cancer and inflammation and discuss potential therapies based on their modulation. Find the latest version: https://jci.me/73940/pdf The Journal of Clinical Investigation REVIEW SERIES: AUTOPHAGY Series Editor: Guido Kroemer The functional and pathologic relevance of autophagy proteases Álvaro F. Fernández and Carlos López-Otín Departamento de Bioquímica y Biología Molecular, Instituto Universitario de Oncología (IUOPA), Universidad de Oviedo, Oviedo, Spain. Autophagy is a well-conserved catabolic process essential for cellular homeostasis. First described in yeast as an adaptive response to starvation, this pathway is also present in higher eukaryotes, where it is triggered by stress signals such as damaged organelles or pathogen infection. -
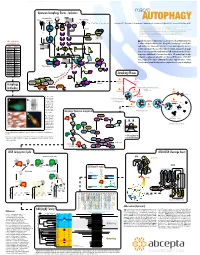
Autophagy Events - Induction Macro
www.abcepta.com Upstream Autophagy Events - Induction macro INS/IGF Environmental stress Wortmannin Glucose Perifosin Amino acids (Leu) Energy stress R LY294002* TKs AUTOPHAGY *1, Eroshkin A1, Mountzouris 2J, Mizushima N3, Hochstrasser M4, Smith JW1, Cuervo AM5 & Klionsky DJ6 Grb cI-PI3K PTEN SOS (1) Burnham Institute for Medical Research, 10901 North Torrey Pines Road, La Jolla, CA 92037 MO25 STRAD IRS1 Ras Nutrients (2) Abcepta Inc., 10320 Camino Santa Fe Ste G, San Diego, CA 92121 LKB1 PIP3 (3) Department of Physiology & Cell Biology, Tokyo Medical and Dental University, Tokyo 113-8519 (4) Molecular Biophysics & Biochemistry, Yale University, New Haven, Connecticut 06520 P Htt-p(Q) (5) Albert Einstein College of Medicine, 1300 Morris Park Avenue, Bronx, NY 10461 AMP P+ Raf P+ Beclin Beclin (6) Life Sciences Institute, University of Michigan, Ann Arbor, Michigan 48109 P P+ (*) corresponding author: [email protected] P P VPS34 Sequestration α MEK1/2 PDK1 VPS15 in HD neurons P+ P+ utophagy is a major self-degradative process in eukaryotic cells, with fundamental roles 100+ additional Ab P P A at www.abgent.com AMPK ERK1/2 Akt/PKB in cellular and organismal homeostasis. Dysregulation of autophagy has recently been AMP Metformin implicated in severe human pathologies such as cancer, neurodegenerative disorders, Cat# Target P P P AICAR P+ P+ P P SC2 SC1 SC2 SC1 SC2 T T T AM1800a LC3 T T P P metabolic and muscular diseases, and viral infections. Structures targeted for autophagic P P P P AP1801a LC3 destruction are sequestered into newly emerging double-membrane vesicles called auto- 14-3-3 AP1802a LC3 GDP GTP P+ Rheb Rheb phagosomes, and delivered for lysosomal degradation. -

Poster Sessions Posters Session 1 Monday November 3, 2014
XXX CONGRESO NACIONAL DE BIOQUÍMICA 2-8 de noviembre de 2014 · Guadalajara, Jal. Poster Sessions Posters Session 1 Monday November 3, 2014. 18:15 – 20:15h SYSTEMS BIOLOGY & BIOINFORMATICS SB-1 Inferring microRNA functions: regulation beyond direct targets. Cesaré Ovando-Vázquez, Roberto Álvarez-Martínez and Cei Abreu-Goodger. LANGEBIO – CINVESTAV – IPN. Irapuato SB-2 Evolution of Codon Usage Bias in Bacteria. Guadalupe Arellano and Luis Delaye. CINVESTAV – IPN Unidad Irapuato SB-3 Adaptive mutations typically increase the performance of metabolism independently from one another. J Abraham Avelar Rivas & Alexander de Luna. LANGEBIO – CINVESTAV – IPN. Irapuato SB-4 Evolutionary codependence as crucial information during LAO protein redesign. Jesús Agustín Banda Vázquez, Rogelio Rodríguez Sotres, Karina Marisol Maya Ramírez y Alejandro Sosa Peinado. Facultad de Medicina. UNAM SB-5 Core and pan-genome analysis of the genus Streptococcus. Hugo Rafael Barajas de la Torre and Luis David Alcaraz. Departamento de Ecología de la Biodiversidad. Instituto de Ecología, UNAM SB-6 Molecular flexibility of a family of periplamic binding proteins for basic aminoacids: LAO-binding protein. Tania Raquel Berrocal Gama, Andrés Escandón Flores, Alejandro Sosa Peinado. Facultad de Medicina. UNAM SB-7 Evaluation of parameters and adequate sampling to measure sea urchin sperm motility. Cecile Bustamante- Gómez, Juan E Sosa-Hernández, Aarón Vazquez Jiménez, Jesús Santana-Solano, Jesús Rodríguez- González and Blanca Estela Galindo. CINVESTAV – IPN. Monterrey SB-8 High-resolution genome-wide aging screens reveal novel molecular mechanisms of lifespan extension by dietary restriction. Sergio E. Campos-Rodríguez & Alexander de Luna. LANGEBIO – CINVESTAV - IPN. Irapuato SB-9 Evolutionary and physiological correlation of redundant gene dosage within the Debaryomyces hansenii genome, a preliminary approach.