Targeted Disruption of the Glutaredoxin 1 Gene Does Not Sensitize Adult Mice to Tissue Injury Induced by Ischemia/Reperfusion and Hyperoxia
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Supplementary Table S4. FGA Co-Expressed Gene List in LUAD
Supplementary Table S4. FGA co-expressed gene list in LUAD tumors Symbol R Locus Description FGG 0.919 4q28 fibrinogen gamma chain FGL1 0.635 8p22 fibrinogen-like 1 SLC7A2 0.536 8p22 solute carrier family 7 (cationic amino acid transporter, y+ system), member 2 DUSP4 0.521 8p12-p11 dual specificity phosphatase 4 HAL 0.51 12q22-q24.1histidine ammonia-lyase PDE4D 0.499 5q12 phosphodiesterase 4D, cAMP-specific FURIN 0.497 15q26.1 furin (paired basic amino acid cleaving enzyme) CPS1 0.49 2q35 carbamoyl-phosphate synthase 1, mitochondrial TESC 0.478 12q24.22 tescalcin INHA 0.465 2q35 inhibin, alpha S100P 0.461 4p16 S100 calcium binding protein P VPS37A 0.447 8p22 vacuolar protein sorting 37 homolog A (S. cerevisiae) SLC16A14 0.447 2q36.3 solute carrier family 16, member 14 PPARGC1A 0.443 4p15.1 peroxisome proliferator-activated receptor gamma, coactivator 1 alpha SIK1 0.435 21q22.3 salt-inducible kinase 1 IRS2 0.434 13q34 insulin receptor substrate 2 RND1 0.433 12q12 Rho family GTPase 1 HGD 0.433 3q13.33 homogentisate 1,2-dioxygenase PTP4A1 0.432 6q12 protein tyrosine phosphatase type IVA, member 1 C8orf4 0.428 8p11.2 chromosome 8 open reading frame 4 DDC 0.427 7p12.2 dopa decarboxylase (aromatic L-amino acid decarboxylase) TACC2 0.427 10q26 transforming, acidic coiled-coil containing protein 2 MUC13 0.422 3q21.2 mucin 13, cell surface associated C5 0.412 9q33-q34 complement component 5 NR4A2 0.412 2q22-q23 nuclear receptor subfamily 4, group A, member 2 EYS 0.411 6q12 eyes shut homolog (Drosophila) GPX2 0.406 14q24.1 glutathione peroxidase -

A State of the Art of Antioxidant Properties of Curcuminoids in Neurodegenerative Diseases
International Journal of Molecular Sciences Review A State of the Art of Antioxidant Properties of Curcuminoids in Neurodegenerative Diseases Serena Silvestro † , Cinzia Sindona †, Placido Bramanti and Emanuela Mazzon * IRCCS Centro Neurolesi “Bonino-Pulejo”, Via Provinciale Palermo, Contrada Casazza, 98124 Messina, Italy; [email protected] (S.S.); [email protected] (C.S.); [email protected] (P.B.) * Correspondence: [email protected]; Tel.: +39-090-60128172 † These authors contributed equally as first author. Abstract: Neurodegenerative diseases represent a set of pathologies characterized by an irreversible and progressive, and a loss of neuronal cells in specific areas of the brain. Oxidative phosphorylation is a source of energy production by which many cells, such as the neuronal cells, meet their energy needs. Dysregulations of oxidative phosphorylation induce oxidative stress, which plays a key role in the onset of neurodegenerative diseases such as Alzheimer’s disease (AD), Parkinson’s disease (PD), and amyotrophic lateral sclerosis (ALS). To date, for most neurodegenerative diseases, there are no resolute treatments, but only interventions capable of alleviating the symptoms or slowing the course of the disease. Therefore, effective neuroprotection strategies are needed. In recent years, natural products, such as curcuminoids, have been intensively explored and studied for their therapeutic potentials in several neurodegenerative diseases. Curcuminoids are, nutraceutical compouns, that owen several therapeutic properties such as anti-oxidant, anti-inflammatory and neuroprotective effects. In this context, the aim of this review was to provide an overview of preclinical and clinical Citation: Silvestro, S.; Sindona, C.; evidence aimed to illustrate the antioxidant effects of curcuminoids in neurodegenerative diseases. -

Role of GSH and Iron-Sulfur Glutaredoxins in Iron Metabolism—Review
molecules Review Role of GSH and Iron-Sulfur Glutaredoxins in Iron Metabolism—Review 1, 1, 1 1 Trnka Daniel y , Hossain Md Faruq y , Jordt Laura Magdalena , Gellert Manuela and Lillig Christopher Horst 2,* 1 Institute for Medical Biochemistry and Molecular Biology, University Medicine, University of Greifswald, 17475 Greifswald, Germany; [email protected] (T.D.); [email protected] (H.M.F.); [email protected] (J.L.M.); [email protected] (G.M.) 2 Christopher Horst Lillig, Institute for Medical Biochemistry and Molecular Biology, University Medicine Greifswald, Ferdinand-Sauerbruch-Straße, 17475 Greifswald, Germany * Correspondence: [email protected]; Tel.: +49-3834-865407; Fax: +49-3834-865402 These authors contributed equally to this work. y Academic Editor: Pál Perjési Received: 29 July 2020; Accepted: 22 August 2020; Published: 25 August 2020 Abstract: Glutathione (GSH) was initially identified and characterized for its redox properties and later for its contributions to detoxification reactions. Over the past decade, however, the essential contributions of glutathione to cellular iron metabolism have come more and more into focus. GSH is indispensable in mitochondrial iron-sulfur (FeS) cluster biosynthesis, primarily by co-ligating FeS clusters as a cofactor of the CGFS-type (class II) glutaredoxins (Grxs). GSH is required for the export of the yet to be defined FeS precursor from the mitochondria to the cytosol. In the cytosol, it is an essential cofactor, again of the multi-domain CGFS-type Grxs, master players in cellular iron and FeS trafficking. In this review, we summarize the recent advances and progress in this field. The most urgent open questions are discussed, such as the role of GSH in the export of FeS precursors from mitochondria, the physiological roles of the CGFS-type Grx interactions with BolA-like proteins and the cluster transfer between Grxs and recipient proteins. -

Urinary Arsenic Methylation Species, Arsenic Methylation Efficiency During Pregnancy and Birth Outcomes
Urinary Arsenic Methylation Species, Arsenic Methylation Efficiency during Pregnancy and Birth Outcomes The Harvard community has made this article openly available. Please share how this access benefits you. Your story matters Citable link http://nrs.harvard.edu/urn-3:HUL.InstRepos:37945565 Terms of Use This article was downloaded from Harvard University’s DASH repository, and is made available under the terms and conditions applicable to Other Posted Material, as set forth at http:// nrs.harvard.edu/urn-3:HUL.InstRepos:dash.current.terms-of- use#LAA Urinary Arsenic Methylation Species, Arsenic Methylation Efficiency during Pregnancy and Birth Outcomes Shangzhi Gao A Dissertation Submitted to the Faculty of The Harvard T.H. Chan School of Public Health in Partial Fulfillment of the Requirements for the Degree of Doctor of Science in the Department of Environmental Health Harvard University Boston, Massachusetts. May, 2018 Dissertation Advisor: Dr. David C. Christiani Shangzhi Gao Urinary Arsenic Methylation Species, Arsenic Methylation Efficiency during Pregnancy and Birth Outcomes Abstract Arsenic exposure from drinking water has been associated with symptoms during pregnancy, as well as negative birth outcomes. The individual’s capability to methylate arsenic modifies the risk of arsenic-related health outcomes. Bangladesh is facing the world’s most severe arsenic crisis, as well as a high prevalence of low birth weight. However, little is known about determinants of arsenic metabolism during pregnancy and its relationship to birth outcomes. Thus, we conducted this study in Bangladesh to identify determinants of arsenic methylation capability during pregnancy, and its relationship with birth outcomes. The study was based on a Bangladesh prospective reproductive cohort (Project Jeebon) of 1,613 pregnant women recruited between 2008 and 2011. -

Variant in the DCTN4 Gene with Wilson Disease
원 저 Journal of Genetic Medicine 2011;8:53-57 • DOI: 10.5734/JGM.2011.8.1.53 53 1) Association of a c.1084A>G (p.Thr362Ala)Variant in the DCTN4 Gene with Wilson Disease Robin Dong-Woo Lee1, 2, Jae-Jung Kim2, Joo-Hyun Kim2, Jong-Keuk Lee2 and Han-Wook Yoo2, 3 1Bel Air High School, Bel Air, MD, USA 2Asan Institute for Life Sciences, University of Ulsan College of Medicine, Seoul, Korea 3Department of Pediatrics, Asan Medical Center Children’s Hospital, University of Ulsan College of Medicine, Seoul, Korea Purpose: Wilson disease is an autosomal recessive disorder which causes excessive copper accumulation in the hepatic region. So far, ATP7B gene is the only disease-causing gene of Wilson disease known to date. However, ATP7B mutations have not been found in ~15% of the patients. This study was performed to identify any causative gene in Wilson disease patients without an ATP7B mutation in either allele. Materials and Methods: The sequence of the coding regions and exon-intron boundaries of the five ATP7B-interacting genes, ATOX1, COMMD1, GLRX, DCTN4, and ZBTB16, were analyzed in the 12 patients with Wilson disease. Results: Three nonsynonymous variants including c.1084A>G (p.Thr362Ala) in the exon 12 of the DCTN4 gene were identified in the patients examined. Among these, only p.Thr362Ala was predicted as possibly damaging protein function by in silico analysis. Examination of allele frequency of c.1084A>G (p.Thr362Ala) variant in the 176 patients with Wilson disease and in the 414 normal subjects revealed that the variant was more prevalent in the Wilson disease patients (odds ratio [OR]=3.14, 95% confidence interval=1.36-7.22, P =0.0094). -

A Potential Therapeutic Role for Angiotensin Converting Enzyme 2 in Human Pulmonary Arterial Hypertension
ERJ Express. Published on June 14, 2018 as doi: 10.1183/13993003.02638-2017 Early View Original article A potential therapeutic role for Angiotensin Converting Enzyme 2 in human pulmonary arterial hypertension Anna R. Hemnes, Anandharajan Rathinasabapathy, Eric A. Austin, Evan L. Brittain, Erica J. Carrier, Xinping Chen, Joshua P. Fessel, Candice D. Fike, Peter Fong, Niki Fortune, Robert E. Gerszten, Jennifer A. Johnson, Mark Kaplowitz, John H. Newman, Robert Piana, Meredith E. Pugh, Todd W. Rice, Ivan M. Robbins, Lisa Wheeler, Chang Yu, James E. Loyd, James West Please cite this article as: Hemnes AR, Rathinasabapathy A, Austin EA, et al. A potential therapeutic role for Angiotensin Converting Enzyme 2 in human pulmonary arterial hypertension. Eur Respir J 2018; in press (https://doi.org/10.1183/13993003.02638-2017). This manuscript has recently been accepted for publication in the European Respiratory Journal. It is published here in its accepted form prior to copyediting and typesetting by our production team. After these production processes are complete and the authors have approved the resulting proofs, the article will move to the latest issue of the ERJ online. Copyright ©ERS 2018 Copyright 2018 by the European Respiratory Society. A potential therapeutic role for Angiotensin Converting Enzyme 2 in human pulmonary arterial hypertension Anna R. Hemnes, MD*1, Anandharajan Rathinasabapathy, PhD*1, Eric A. Austin, MD, MSCI2, Evan L. Brittain, MD, MSCI3, Erica J. Carrier, PhD1, Xinping Chen, PhD1, Joshua P. Fessel, MD, PhD1, Candice D. Fike, MD2, Peter Fong, MD3, Niki Fortune1, Robert E. Gerszten, MD4, Jennifer A. Johnson, MD1, Mark Kaplowitz2, John H. -
Up- and Down-Regulated Molecules Comparing Coiled Versus
On-line Table 1: Up- and down-regulated molecules comparing coiled versus untreated aneurysms, determined by IPAa Gene Main Function Determined by Gene Fold False Discovery Direction of Gene Name Description Ontology Change P Value Rate (q Value) Expression ABCB6 ATP-binding cassette, sub-family B (MDR/ Integral component of mitochondrial outer 2.541 4.28E-05 5.90E-04 Up-regulated TAP), member 6 (Langereis blood group) membrane ABCC1 ATP-binding cassette, sub-family C (CFTR/ ATPase activity, coupled to transmembrane 3.132 4.94E-10 6.17E-08 Up-regulated MRP), member 1 movement of substances ACOT11 Acyl-coa thioesterase 11 Carboxylic ester hydrolase activity 3.973 2.58E-04 2.42E-03 Up-regulated ADAM10 ADAM metallopeptidase domain 10 PMA-inducible membrane protein 3.425 1.33E-04 1.44E-03 Up-regulated ectodomain proteolysis ADAM8 ADAM metallopeptidase domain 8 Positive regulation of tumor necrosis factor 3.417 1.42E-13 6.74E-11 Up-regulated (ligand) superfamily member 11 production ADAMTS4 ADAM metallopeptidase with Proteinaceous extracellular matrix 2.134 2.49E-08 1.52E-06 Up-regulated thrombospondin type 1 motif, 4 ADH4 Alcohol dehydrogenase 4 (class II), pi Oxidoreductase activity, acting on the Ϫ2.657 4.44E-05 6.04E-04 Down-regulated polypeptide aldehyde or oxo group of donors, NAD or NADP as acceptor ADORA2B Adenosine A2b receptor Positive regulation of chronic inflammatory 2.056 1.95E-05 3.22E-04 Up-regulated response to non-antigenic stimulus AK4 Adenylate kinase 4 Nucleoside triphosphate adenylate kinase 2.074 1.96E-08 1.25E-06 Up-regulated -

Copper/Iron/Ceruloplasmin/Magnesium
Date of Analysis: 2/7/2019 GVA Version: 558 Client: Jane Sample Copper/Iron/Ceruloplasmin/Magnesium Clearly iron is an important mineral needed for many functions. However, if iron is in excess or it is not properly chaperoned through the body, it can become a very dangerous free radical. Because iron has a limited capacity to be excreted by the body, it can easily build up in organs like the liver, heart, and pancreas. This is potentially problematic because iron is a potent oxidizer that can damage body tissues. This section of the pyramid covers genes such as the HFE gene (which could cause higher levels of iron absorption), the SLC4A1 and SLC41A2 gene (which transports magnesium), and the SLC40A1 gene (which makes ferroportin). Ferroportin is the only iron exporter that could keep iron in the cell. If this happens, there is a potential through the Fenton Reaction to create hydroxyl radicals. If there is excess iron, it is possible that through the Fenton Reaction, iron combines with hydrogen peroxide to make very dangerous hydroxyl radicals. As shown in this diagram, excess iron will combine with hydrogen peroxide to make hydroxyl radicals. To make sure that iron is chaperoned properly in the body, there is a need for ceruloplasmin. Some of the cofactors needed to make ceruloplasmin are copper, and vitamin A. When there are variants in the BCMO1 gene, there may be impaired beta-carotene to vitamin A conversion. Because of this, ceruloplasmin levels may be low. If you see this pattern it may be valuable to measure ceruloplasmin levels. -

Autocrine IFN Signaling Inducing Profibrotic Fibroblast Responses By
Downloaded from http://www.jimmunol.org/ by guest on September 23, 2021 Inducing is online at: average * The Journal of Immunology , 11 of which you can access for free at: 2013; 191:2956-2966; Prepublished online 16 from submission to initial decision 4 weeks from acceptance to publication August 2013; doi: 10.4049/jimmunol.1300376 http://www.jimmunol.org/content/191/6/2956 A Synthetic TLR3 Ligand Mitigates Profibrotic Fibroblast Responses by Autocrine IFN Signaling Feng Fang, Kohtaro Ooka, Xiaoyong Sun, Ruchi Shah, Swati Bhattacharyya, Jun Wei and John Varga J Immunol cites 49 articles Submit online. Every submission reviewed by practicing scientists ? is published twice each month by Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts http://jimmunol.org/subscription Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html http://www.jimmunol.org/content/suppl/2013/08/20/jimmunol.130037 6.DC1 This article http://www.jimmunol.org/content/191/6/2956.full#ref-list-1 Information about subscribing to The JI No Triage! Fast Publication! Rapid Reviews! 30 days* Why • • • Material References Permissions Email Alerts Subscription Supplementary The Journal of Immunology The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2013 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. This information is current as of September 23, 2021. The Journal of Immunology A Synthetic TLR3 Ligand Mitigates Profibrotic Fibroblast Responses by Inducing Autocrine IFN Signaling Feng Fang,* Kohtaro Ooka,* Xiaoyong Sun,† Ruchi Shah,* Swati Bhattacharyya,* Jun Wei,* and John Varga* Activation of TLR3 by exogenous microbial ligands or endogenous injury-associated ligands leads to production of type I IFN. -

The Involvement of Anti-Oxidative Response and Mitochondrial Dynamics in the Pathogenesis of Friedreich's Ataxia: Relevance T
The Involvement of Anti-Oxidative Response and Mitochondrial Dynamics in the Pathogenesis of Friedreich’s Ataxia: Relevance to the Development of Future Therapeutics Shannon Chiang B. Med. Sci. Grad. Dip. Sc. Doctor of Philosophy (Medicine) Discipline of Pathology Faculty of Medicine and Health The University of Sydney 2019 A thesis submitted in fulfilment of the requirements for the degree of Doctor of Philosophy in the Faculty of Medicine and Health at the University of Sydney ACKNOWLEDGEMENTS I would like to first thank my supervisor, Prof. Des Richardson, for you have given me this extraordinary opportunity to pursue my interest in scientific research, allowing me to be part of your laboratory, and to fulfil my PhD studies. Thank you for believing in me. I am grateful that you have accepted me. Your knowledge and insight have helped me over the years, and I appreciate all the efforts and time you have spent to guide me through this journey. Thank you for your many hours of proofreading and writing assistance in preparing the articles and thesis that have been the backbone of this contribution. To my associate supervisor and friend, Dr. Michael Huang, thank you very much for all the help, support, and guidance you have given me. You are a wonderful mentor and you have taught me many valuable lessons. Thank you for being so patient, caring, and supportive. I am grateful to have been your student, and I enjoy our partnership. As you have once said, ‘every moment is an opportunity to learn something new’, I will continue to do so. -
PDF-Document
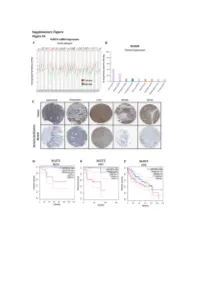
Figure S1: NUDT5 is indicative of poor prognosis and aggressive cancer growth. (A) NUDT5 mRNA expression across full TGCA database, data for tumour (red) and normal (green) tissue from the same tissue origin are shown using TGCA patient datasets with GEPIA interactive webserver [61]. Datasets where NUDT5 mRNA levels are elevated in tumour versus normal as highlighted in red, significantly higher in normal versus tumour are marked in green. Abbreviations of the tumour datasets are given in Table S1. (B) The percentage of positive staining for NUDT5 protein expression in cancer datasets (Human Protein Atlas database [62]). (C) Representative histological staining of NUDT5 in tumour versus normal patient samples from colorectal, pancreatic, liver, breast and renal samples (Human Protein Atlas database [62]). Metadata regarding each of the images are given in Table S2. Kaplan Meyer survival curves stratifying patients based on NUDT5 mRNA expression in (D) Kidney chromophobe (KICH). (E) Adrenalcorticocal carcinoma (ACC). (F) Liver hepatocellular carcinoma (LIHG) (G) Brain lower grade carcinoma (LGG) (H) Kidney renal clear cell carcinoma (KIRC) (I) Kidney renal papillary cell carcinoma (KIRP) were carried out using TGCA database with GEPIA interactive webserver [61]. (J) Recurrence and metastasis analysis in NUDT5 mRNA high expressing patient cohorts from BRCA dataset shown in Figure S1A. (K) Full western blot of NUDT5, PARP1 and FLAG protein levels in NUDT5RES and NUDT5KD cell clones. Figure 2. Analysis of the gene expression changes in 3D compared to 2D culture conditions. (A) PCA analysis of all RNA samples (Table S3) from T47D cells cultured in 2D and 3D conditions. (B) PCA analysis of RNA samples from T47D cells highlighted based on culture conditions. -

Table S1. 103 Ferroptosis-Related Genes Retrieved from the Genecards
Table S1. 103 ferroptosis-related genes retrieved from the GeneCards. Gene Symbol Description Category GPX4 Glutathione Peroxidase 4 Protein Coding AIFM2 Apoptosis Inducing Factor Mitochondria Associated 2 Protein Coding TP53 Tumor Protein P53 Protein Coding ACSL4 Acyl-CoA Synthetase Long Chain Family Member 4 Protein Coding SLC7A11 Solute Carrier Family 7 Member 11 Protein Coding VDAC2 Voltage Dependent Anion Channel 2 Protein Coding VDAC3 Voltage Dependent Anion Channel 3 Protein Coding ATG5 Autophagy Related 5 Protein Coding ATG7 Autophagy Related 7 Protein Coding NCOA4 Nuclear Receptor Coactivator 4 Protein Coding HMOX1 Heme Oxygenase 1 Protein Coding SLC3A2 Solute Carrier Family 3 Member 2 Protein Coding ALOX15 Arachidonate 15-Lipoxygenase Protein Coding BECN1 Beclin 1 Protein Coding PRKAA1 Protein Kinase AMP-Activated Catalytic Subunit Alpha 1 Protein Coding SAT1 Spermidine/Spermine N1-Acetyltransferase 1 Protein Coding NF2 Neurofibromin 2 Protein Coding YAP1 Yes1 Associated Transcriptional Regulator Protein Coding FTH1 Ferritin Heavy Chain 1 Protein Coding TF Transferrin Protein Coding TFRC Transferrin Receptor Protein Coding FTL Ferritin Light Chain Protein Coding CYBB Cytochrome B-245 Beta Chain Protein Coding GSS Glutathione Synthetase Protein Coding CP Ceruloplasmin Protein Coding PRNP Prion Protein Protein Coding SLC11A2 Solute Carrier Family 11 Member 2 Protein Coding SLC40A1 Solute Carrier Family 40 Member 1 Protein Coding STEAP3 STEAP3 Metalloreductase Protein Coding ACSL1 Acyl-CoA Synthetase Long Chain Family Member 1 Protein