Therapeutic Use of the Ketogenic Diet in Refractory Epilepsy: What We Know and What Still Needs to Be Learned
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

01 Excipients Prelims 1..9
Triolein 757 and tablets). Included in the Canadian List of Acceptable Non- 3 Steurnagel CR. Latex emulsions for controlled drug delivery. McGinity medicinal Ingredients. JW, ed. Aqueous Polymeric Coatings for Pharmaceutical Dosage Forms. New York: Marcel Dekker, 1989; 1–61. 4 Gutierrez-Rocca JC, McGinity JW. Influence of aging on the physical– 17 Related Substances mechanical properties of acrylic resin films cast from aqueous Acetyltributyl citrate; acetyltriethyl citrate; tributyl citrate. dispersions and organic solutions. Drug Dev Ind Pharm 1993; 19(3): 315–332. 5 Liu J, Williams R. Properties of heat-humidity cured cellulose acetate 18 Comments phthalate free films. Eur J Pharm Sci 2002; 17(1–2): 31–41. 6 Lewis RJ, ed. Sax’s Dangerous Properties of Industrial Materials, 11th A specification for triethyl citrate is contained in the Food (7) edn. New York: Wiley, 2004; 3546. Chemicals Codex (FCC). 7 Food Chemicals Codex, 6th edn. Bethesda, MD: United States The EINECS number for triethyl citrate is 201-070-7. The Pharmacopeia, 2008; 988. PubChem Compound ID (CID) for triethyl citrate is 6506. 20 General References 19 Specific References Vertellus Specialties Inc. Technical data sheet: Citroflex 2, 2007. 1 Gutierrez-Rocca JC, McGinity JW. Influence of water soluble and insoluble plasticizers on the physical and mechanical properties of 21 Author acrylic resin copolymers. Int J Pharm 1994; 103: 293–301. J Teckoe. 2 Lehmann K. Chemistry and application properties of polymethacrylate coating systems. McGinity JW, ed. Aqueous Polymeric Coatings for Pharmaceutical Dosage Forms. New York: Marcel Dekker, 1989; 153– 22 Date of Revision 245. 24 February 2009. Triolein 1 Nonproprietary Names 6 Functional Category None adopted. -

Intravenous Treatment with a Long-Chain Omega-3 Lipid Emulsion Provides Neuroprotection in a Murine Model of Ischemic Stroke – a Pilot Study
RESEARCH ARTICLE Intravenous Treatment with a Long-Chain Omega-3 Lipid Emulsion Provides Neuroprotection in a Murine Model of Ischemic Stroke ± A Pilot Study Dirk Berressem1*, Konrad Koch1, Nicole Franke1, Jochen Klein1, Gunter P. Eckert1,2 1 Goethe-University of Frankfurt, Department of Pharmacology, Germany, 2 Justus-Liebig-University Giessen, Institute of Nutritional Sciences, Germany * [email protected] a11111 Abstract Single long-chain omega-3 fatty acids (e.g. docosahexaenoic acid (DHA) or eicosapentae- noic acid (EPA)) are known for their neuroprotective properties associated with ischemic stroke. This pilot study aimed to test the effectiveness of an acute treatment with a long- OPEN ACCESS chain omega-3 lipid emulsion (Omegaven 10%®, OGV) that contains fish oil (DHA 18 mg/ Citation: Berressem D, Koch K, Franke N, Klein J, Eckert GP (2016) Intravenous Treatment with a ml; EPA 21 mg/ml) and α-tocopherol (0.2 mg/ml) in a transient middle cerebral artery occlu- Long-Chain Omega-3 Lipid Emulsion Provides sion (MCAO) model of ischemic stroke in mice. For this purpose, female CD-1 mice were Neuroprotection in a Murine Model of Ischemic anesthetized and subjected to 90 minutes of MCAO. To reflect a clinically relevant situation Stroke ± A Pilot Study. PLoS ONE 11(11): for an acute treatment, either after induction of stroke or after reperfusion, a single dose of e0167329. doi:10.1371/journal.pone.0167329 OGV was injected intravenously into the tail vein (5 ml/kg b.w.). A neurological severity Editor: Muzamil Ahmad, Indian Institute of score was used to assess motor function and neurological outcome. -

Triheptanoin for Glucose Transporter Type I Deficiency (G1D) Modulation of Human Ictogenesis, Cerebral Metabolic Rate, and Cognitive Indices by a Food Supplement
Research Original Investigation Triheptanoin for Glucose Transporter Type I Deficiency (G1D) Modulation of Human Ictogenesis, Cerebral Metabolic Rate, and Cognitive Indices by a Food Supplement Juan M. Pascual, MD, PhD; Peiying Liu, PhD; Deng Mao, BS; Dorothy I. Kelly, MA; Ana Hernandez, MS; Min Sheng, PhD; Levi B. Good, PhD; Qian Ma, MD, PhD; Isaac Marin-Valencia, MD, MS; Xuchen Zhang, MD; Jason Y. Park, MD, PhD; Linda S. Hynan, PhD; Peter Stavinoha, PhD; Charles R. Roe, MD; Hanzhang Lu, PhD Supplemental content at IMPORTANCE Disorders of brain metabolism are multiform in their mechanisms and jamaneurology.com manifestations, many of which remain insufficiently understood and are thus similarly treated. Glucose transporter type I deficiency (G1D) is commonly associated with seizures and with electrographic spike-waves. The G1D syndrome has long been attributed to energy (ie, adenosine triphosphate synthetic) failure such as that consequent to tricarboxylic acid (TCA) cycle intermediate depletion. Indeed, glucose and other substrates generate TCAs via anaplerosis. However, TCAs are preserved in murine G1D, rendering energy-failure inferences premature and suggesting a different hypothesis, also grounded on our work, that consumption of alternate TCA precursors is stimulated and may be detrimental. Second, common ketogenic diets lead to a therapeutically counterintuitive reduction in blood glucose available to the G1D brain and prove ineffective in one-third of patients. OBJECTIVE To identify the most helpful outcomes for treatment evaluation and to uphold (rather than diminish) blood glucose concentration and stimulate the TCA cycle, including anaplerosis, in G1D using the medium-chain, food-grade triglyceride triheptanoin. DESIGN, SETTING, AND PARTICIPANTS Unsponsored, open-label cases series conducted in an academic setting. -

6 Minute Walk Results
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date r i 1 /1 i 22 December 2011 (22.12.2011) » 2U1 1/159634ft Al (51) International Patent Classification: (81) Designated States (unless otherwise indicated, for every A61K 31/23 (2006.01) A61P 3/00 (2006.01) kind of national protection available): AE, AG, AL, AM, AO, AT, AU, AZ, BA, BB, BG, BH, BR, BW, BY, BZ, (21) International Application Number: CA, CH, CL, CN, CO, CR, CU, CZ, DE, DK, DM, DO, PCT/US201 1/040234 DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, (22) International Filing Date: HN, HR, HU, ID, JL, IN, IS, JP, KE, KG, KM, KN, KP, 13 June 201 1 (13.06.201 1) KR, KZ, LA, LC, LK, LR, LS, LT, LU, LY, MA, MD, ME, MG, MK, MN, MW, MX, MY, MZ, NA, NG, NI, (25) Filing Language: English NO, NZ, OM, PE, PG, PH, PL, PT, RO, RS, RU, SC, SD, (26) Publication Language: English SE, SG, SK, SL, SM, ST, SV, SY, TH, TJ, TM, TN, TR, TT, TZ, UA, UG, US, UZ, VC, VN, ZA, ZM, ZW. (30) Priority Data: 61/354,472 14 June 2010 (14.06.2010) US (84) Designated States (unless otherwise indicated, for every 13/159,329 13 June 201 1 (13.06.201 1) US kind of regional protection available): ARIPO (BW, GH, GM, KE, LR, LS, MW, MZ, NA, SD, SL, SZ, TZ, UG, (71) Applicant (for all designated States except US): BAY¬ ZM, ZW), Eurasian (AM, AZ, BY, KG, KZ, MD, RU, TJ, LOR RESEARCH INSTITUTE [US/US]; 33 10 Live TM), European (AL, AT, BE, BG, CH, CY, CZ, DE, DK, Oak Street, Suite 501, Dallas, TX 75204 (US). -

Chemoprevention in Kidney Cancer by Madhur Nayan
Chemoprevention in Kidney Cancer by Madhur Nayan A thesis submitted in conformity with the requirements for the degree of Doctor of Philosophy in Clinical Epidemiology, Graduate Department of Health Policy, Management, and Evaluation, in the University of Toronto © Copyright by Madhur Nayan, 2017 THESIS ABSTRACT Thesis Title: Chemoprevention in kidney cancer Degree: Doctor of Philosophy (PhD) in Clinical Epidemiology Year of Convocation: 2017 Student: Madhur Nayan Graduate Department: Health Policy, Management and Evaluation University: University of Toronto Background: This thesis is a composition of three studies that explore the role of statins in kidney cancer. Furthermore, I evaluate the potential for different interpretations from the same data depending on the method of classifying medication use. Methods: The first study was a population-based case-control study evaluating the association of statin use with risk of incident kidney cancer. The second study was a systematic review and meta-analysis reviewing the current evidence relating statins with kidney cancer survival outcomes. The final study was a population-based cohort study evaluating the association of statin use with survival. In the observational studies, I used fractional polynomials for the primary analysis to allow for a non-linear relationship between cumulative exposure and the risk of the outcome. I also compared risk estimates obtained by different methods of classifying medication exposure. Results: The population-based case-control study included 10,377 incident cases of kidney cancer and 35,939 matched controls. Increasing cumulative use of statins was not associated with kidney cancer risk. I identified 12 studies for inclusion in the systematic review and meta- analysis and found that statin use was significantly associated with markedly improved cancer- specific and overall survival. -
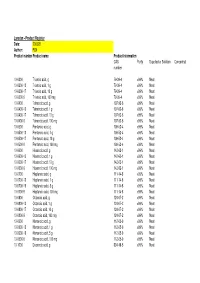
Larodan - Product Register Date: 200528 Author: FLN Product Number Product Name Product Information CAS Purity Supplied As Solution Concentration Number
Larodan - Product Register Date: 200528 Author: FLN Product number Product name Product information CAS Purity Supplied as Solution Concentration number 10-0300 Trianoic acid, g 79-09-4 >99% Neat 10-0300-13 Trianoic acid, 1 g 79-09-4 >99% Neat 10-0300-17 Trianoic acid, 10 g 79-09-4 >99% Neat 10-0300-9 Trianoic acid, 100 mg 79-09-4 >99% Neat 10-0400 Tetranoic acid, g 107-92-6 >99% Neat 10-0400-13 Tetranoic acid, 1 g 107-92-6 >99% Neat 10-0400-17 Tetranoic acid, 10 g 107-92-6 >99% Neat 10-0400-9 Tetranoic acid, 100 mg 107-92-6 >99% Neat 10-0500 Pentanoic acid, g 109-52-4 >99% Neat 10-0500-13 Pentanoic acid, 1 g 109-52-4 >99% Neat 10-0500-17 Pentanoic acid, 10 g 109-52-4 >99% Neat 10-0500-9 Pentanoic acid, 100 mg 109-52-4 >99% Neat 10-0600 Hexanoic acid, g 142-62-1 >99% Neat 10-0600-13 Hexanoic acid, 1 g 142-62-1 >99% Neat 10-0600-17 Hexanoic acid, 10 g 142-62-1 >99% Neat 10-0600-9 Hexanoic acid, 100 mg 142-62-1 >99% Neat 10-0700 Heptanoic acid, g 111-14-8 >99% Neat 10-0700-13 Heptanoic acid, 1 g 111-14-8 >99% Neat 10-0700-16 Heptanoic acid, 5 g 111-14-8 >99% Neat 10-0700-9 Heptanoic acid, 100 mg 111-14-8 >99% Neat 10-0800 Octanoic acid, g 124-07-2 >99% Neat 10-0800-13 Octanoic acid, 1 g 124-07-2 >99% Neat 10-0800-17 Octanoic acid, 10 g 124-07-2 >99% Neat 10-0800-9 Octanoic acid, 100 mg 124-07-2 >99% Neat 10-0900 Nonanoic acid, g 112-05-0 >99% Neat 10-0900-13 Nonanoic acid, 1 g 112-05-0 >99% Neat 10-0900-16 Nonanoic acid, 5 g 112-05-0 >99% Neat 10-0900-9 Nonanoic acid, 100 mg 112-05-0 >99% Neat 10-1000 Decanoic acid, g 334-48-5 >99% Neat -

Fatty Acids & Derivatives
Conditions of Sale Validity The Conditions of Sale apply to the written text in this Catalogue superseding earlier texts related to such conditions. Intention of Use Our products are intended for research purposes only. Prices See under Order Information. All prices in this catalogue are net prices in Euro, ex works. Taxes, shipping costs or other external costs demanded by the buyer are invoiced. Delivery See under Order Information – shipping terms. Payment terms Payment terms are normally net 30 days. Deductions are not accepted unless we have issued a credit note. We accept payment by credit card (Visa/ Mastercard), bank transfer (wiring) or by cheque. If payment by cheque we will add a bank fee to our invoice. Complaints Complaints about a product or products must be made inside 30 days from the invoice date. All claims must specify batch (lot) and invoice numbers. Return of goods will not be accepted unless authorized by us. Insurance Insurance will not be made unless otherwise instructed. Delays We cannot accept compensation claims due to delays or non-deliveries. We reserve us the right to withdraw from delivery due to long term shortage of starting materials, production breakdown or other circumstances beyond our control. Warranty and All products in this catalogue are warranted to be free of defects and in Compensation Claims accordance with given specifications. If this warranty does not comply with specifications, any indemnities will be limited to not exceed the price paid for the goods. Acceptance Placing of an order implies acceptance of our conditions of sale. We accept credit cards (Visa/ Mastercard) www.larodan.se [email protected] +46 40 16 41 55 1 Ordering Information You can easily order from Larodan – please contact our local distributor or us by phone, e-mail, fax or letter. -

(12) Patent Application Publication (10) Pub. No.: US 2009/0221978 A1 Gatto Et Al
US 20090221978A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2009/0221978 A1 Gatto et al. (43) Pub. Date: Sep. 3, 2009 (54) ABSORBENT ARTICLE WITH LOTION Publication Classification COMPRISINGAPOLY PROPYLENE GLYCOL (51) Int. Cl. MATERAL A6F 3/15 (2006.01) (76) Inventors: Joseph Anthony Gatto, Redondo (52) U.S. Cl. ................ 604/367; 604/385.23; 604/385.01 Beach, CA (US); Brent Taylor (57) ABSTRACT Ginn, Monroe, OH (US); Robert An absorbent article, such as a catamenial device, comprises Ya-lin Pan, Cincinnati, OH (US) a liquid pervious topsheet, the topsheet having an inner Sur face oriented toward the interior of the absorbent article and Correspondence Address: an outer surface oriented toward the skin of the wearer when THE PROCTER & GAMBLE COMPANY the absorbent article is being worn. The absorbent article Global Legal Department - IP includes a backsheet joined to the topsheet, the backsheet Sycamore Building - 4th Floor, 299 East Sixth having an inner surface oriented toward the interior of the Street absorbent article and an outer surface oriented toward the CINCINNATI, OH 45202 (US) garment of the wearer when the absorbent article is being worn. The absorbent article includes an absorbent core dis (21) Appl. No.: 12/369,922 posed between the topsheet and the backsheet, the absorbent core having an inner Surface oriented toward the skin of the (22) Filed: Feb. 12, 2009 wearer when the absorbent article is being worn and an outer surface oriented toward the garment of the wearer when the absorbent article is being worn. The absorbent article Related U.S. -

Amended Safety Assessment of Triglycerides As Used in Cosmetics
Amended Safety Assessment of Triglycerides as Used in Cosmetics Status: Re-Review for Panel Review Release Date: March 17, 2017 Panel Meeting Date: April 10-11, 2017 The 2017 Cosmetic Ingredient Review Expert Panel members are: Chairman, Wilma F. Bergfeld, M.D., F.A.C.P.; Donald V. Belsito, M.D.; Ronald A. Hill, Ph.D.; Curtis D. Klaassen, Ph.D.; Daniel C. Liebler, Ph.D.; James G. Marks, Jr., M.D., Ronald C. Shank, Ph.D.; Thomas J. Slaga, Ph.D.; and Paul W. Snyder, D.V.M., Ph.D. The CIR Director is Lillian J. Gill, D.P.A. This safety assessment was prepared by Monice M. Fiume, Assistant Director/Senior Scientific Analyst/Writer, and Bart Heldreth, Chemist. © Cosmetic Ingredient Review 1620 L Street, NW, Suite 1200 ♢ Washington, DC 20036-4702 ♢ ph 202.331.0651 ♢ fax 202.331.0088 ♢ [email protected] Commitment & Credibility since 1976 Memorandum To: CIR Expert Panel Members and Liaisons From: Monice M. Fiume MMF Assistant Director/Senior Scientific Analyst Date: March 17, 2017 Subject: Amended Safety Assessment of Triglycerides as Used in Cosmetics Enclosed is the Safety Assessment of Triglycerides as Used in Cosmetics. (It is identified as trygly042017rep in the pdf document.) This is a re-review that is being initiated in accord with CIR’s Procedures to reassess previously- reviewed conclusions after a period of 15 years. In 2000, the Panel published a safety assessment of Trihydroxystearin with the conclusion, “Based on the available animal and clinical data, which included summary data from the CIR safety assessments of Hydroxystearic Acid and Glyceryl Stearate and Glyceryl Stearate SE, the Panel concluded that Trihydroxystearin is safe as used in cosmetics.” In 2015, the Panel re-evaluated the safety of Hydroxystearic Acid and Glyceryl Stearate and Glyceryl Stearate SE, reaffirming that Hydroxystearic Acid is safe as a cosmetic ingredient in the present practices of use and concluding that Glyceryl Stearate and Glyceryl Stearate SE are safe in the present practices of use and concentration. -

NCT Number: 02036853 Sponsor: Cook Children’S Medical Center Primary Investigator: Adrian Lacy, MD
Study Title: An Open-Label Trial of Triheptanoin in Patients with Glucose Transporter Type-1 Deficiency Syndrome(GLUT1 DS) Investigational Product: Triheptanoin Indication: Glucose Transporter Type-1 Deficiency Syndrome (GLUT1DS) IND Number: 120505 NCT Number: 02036853 Sponsor: Cook Children’s Medical Center Primary Investigator: Adrian Lacy, MD Cook Children’s Study Number: 2013-RNEU -001 Protocol Version Date: Version 4.1, 23-Mar-2018 Company: Ultragenyx Pharmaceutical, Inc. Study Site: Cook Children’s Medical Center, 801 7th Ave, Fort Worth, TX 76104 Phase of Development: Phase 2 IRB NUMBER: 2013-034 IRB APPROVAL DATE: 04/04/2019 IRB EXPIRATION DATE: 04/03/2020 Protocol Number: 2013-NEUR-001 Version: 4.1 23-Mar-2018 TABLE OF CONTENTS SECTION PAGE 1.0 STUDY OBJECTIVES 4 1.1 Primary Objectives 4 1.2 Secondary Objectives 4 2.0 BACKGROUND AND SIGNIFICANCE 4-7 2.1 Disease Background 4 2.2 Supporting Previous Studies 4 2.3 Protocol Rationale 7 3.0 STUDY DESIGN 7-8 3.1 Study Population 7 3.2 Number of Subjects Planned 7 3.3 Study Timeline 8 4.0 SELECTION AND ENROLLMENT OF SUBJECTS 9-10 4.1 Inclusion Criteria 9 4.2 Exclusion Criteria 9 4.3 Consent Procedures 10 5.0 METHODS 10-13 5.1 Recruitment of Subjects 10 5.2 Sources of Research Material 10 5.3 Duration of Treatment 11 5.4 Study Procedures 11 6.0 OBSERVATIONS AND MEASUREMENTS 14-16 6.1 Vineland Adaptive Behavior Scales, Second Edition 14 6.2 Barry-Albright Dystonia Scale 14 6.3 The Pediatric Quality of Life Inventory Scale 15 6.4 Columbia Suicide Severity Rating Scale 15 6.5 Tracking Diary -

Redalyc.Hepatic Fatty Acid Profile of Rats Fed a Triheptanoin-Based
Nutrición Hospitalaria ISSN: 0212-1611 info@nutriciónhospitalaria.com Grupo Aula Médica España Vieira de Melo, Ingrid Sofia; da Rocha Ataide, Terezinha; Lima de Oliveira, Suzana; Bezerra Bueno, Nassib; Duarte de Freitas, Johnnatan; Goulart Sant’Ana, Antônio Euzébio Hepatic fatty acid profile of rats fed a triheptanoin-based ketogenic diet Nutrición Hospitalaria, vol. 32, núm. 1, 2015, pp. 265-269 Grupo Aula Médica Madrid, España Available in: http://www.redalyc.org/articulo.oa?id=309239661038 How to cite Complete issue Scientific Information System More information about this article Network of Scientific Journals from Latin America, the Caribbean, Spain and Portugal Journal's homepage in redalyc.org Non-profit academic project, developed under the open access initiative Nutr Hosp. 2015;32(1):265-269 ISSN 0212-1611 • CODEN NUHOEQ S.V.R. 318 Original / Investigación animal Hepatic fatty acid profile of rats fed a triheptanoin-based ketogenic diet Ingrid Sofia Vieira de Melo1, Terezinha da Rocha Ataide2, Suzana Lima de Oliveira2, Nassib Bezerra Bueno2, Johnnatan Duarte de Freitas3 and Antônio Euzébio Goulart Sant’Ana4 1Departamento de Agroindústria, Instituto Federal de Educação Ciência e Tecnologia de Alagoas, Murici, AL. 2Laboratório de Nutrição Experimental, Faculdade de Nutrição, Universidade Federal de Alagoas, Maceió, AL. 3Departamento de Tecnologia de Alimentos, Instituto Federal de Educação Ciência e Tecnologia de Alagoas, Maceió - AL. 4Laboratório de Recursos Naturais, Instituto de Química e Biotecnologia, Universidade Federal de Alagoas, Maceió, AL, Brasil. Abstract PERFIL DE ÁCIDOS GRASOS DE HÍGADOS DE RATONES ALIMENTADOS CON UNA DIETA Objective: the aim of this study was to evaluate the in- CETOGÉNICA BASADA EN TRIHEPTANOÍNA fluence of consumption of a ketogenic diet supplemented with triheptanoin, a medium-chain anaplerotic triacyl- glycerol, on the liver fatty acid profile of Wistar rats. -

(MCT; C8) and Triheptanoin (C7) in Patients With
! A Comparison of the Digestion and Absorption of Medium-Chain Triglyceride (MCT; C8) and Triheptanoin (C7) in Patients with Long-Chain Fatty Acid Oxidation Disorders By Kayla K. Guillory A THESIS Presented to the Graduate Programs in Human Nutrition and the Oregon Health & Science University School of Medicine in partial fulfillment of the requirements for the degree of Master of Science in Clinical Nutrition June 2016 ! ! June 2016 School of Medicine Oregon Health Science University Certificate of Approval This is certifying that the Master’s thesis of Kayla K. Guillory has been approved Mentor/ Advisor Member Member ! ! Table of Contents Page List of Figures iv List of Tables v Abbreviations vi Units of Measure ix Acknowledgments x Abstract xi Chapter 1: Specific Aims 1 Chapter 2: Background 4 Chapter 3: Research Design 19 Chapter 4: Methods 23 Chapter 5: Statistical Analysis 24 Chapter 6: Results 35 Chapter 7: Discussion 57 References 62 Appendix 1: Free Fatty Acid Assay Procedure 65 Appendix 2: Evidence Table 66 i ! ! List of Figures Figure # Title Page Figure 1: β-Oxidation Cycle 8 Figure 2: Anaplerotic Mechanism of Triheptanoin (C7) 12 Figure 3: Macronutrient Composition of Standardized Meal 38 Figure 4: Plasma Free Fatty Acids 41 Figure 5: Medium-Chain Fatty Acid Profiles 44 Figure 6: Total Change in Plasma C8 and C7 after Mixed Meal 46 Figure 7: Long-Chain Fatty Acid Profiles 47 Figure 8: Cumulative Recovery 49 Figure 9: Ketones 52 Figure 10: Acylcarnitines 55 ii ! ! List of Tables Table # Title Page Table 1: Overall Study Design