COMPLEX SYSTEMS BIOLOGY of MAMMALIAN CELL CYCLE SIGNALING in CANCER by JAYANT AVVA Submitted in Partial Fulfillment of the Requi
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

COMPLEX SYSTEMS BIOLOGY and HEGEL's PHILOSOPHY Kazuyuki
COMPLEX SYSTEMS BIOLOGY AND HEGEL’S PHILOSOPHY Kazuyuki Ikko Takahashi KandaSurugadai 1-1, Chiyoda, Tokyo 101-8301, Meiji University ABSTRACT In this study I will argue that Hegel’s philosophy has similarity to the self- organization theories of Prigogine and Kauffman and complex systems biology of Kaneko, and is therefore an idea in advance of its times. In The Philosophy of Nature, Hegel’s interest is in how nature evolves through the mechanism of self-organization. He was writing before Darwin proposed the theory of evolution, and his dialectic is aimed at analyzing and describing development in the logical sense. The important feature of this work is their analysis of the fundamental structures by which order is generated. Hegel struggled to produce the concept of life from that of matter. He proposed that matter should develop into organism, but only in a logical sense. Nature itself is a system of producing spontaneous order through the random motion of the contingent. Then Hegel tackles living things. He would like to say that the basis of life is the non-equilibrium self-referential structure. In more modern terminology, we could interpret this as meaning that the first organism emerged from interaction between high polymers. Living creatures exhibit flexibility and plasticity through fluctuations in these elements. Complex systems biology uses a dynamical systems approach to explain how living things acquire diversity, stability and spontaneity. First, simple single-celled organisms arose through interactions between proteins and nucleic acids. These are the archae-bacteria in modern terminology. Next, the development of eukaryote cells from the prokaryotes is explained by symbiogenesis or endosymbiotic theory. -

Complex Systems Analysis of Arrested Neural Cell Differentiation During Development and Analogous Cell Cycling Models in Carcinogenesis
[04-01-2012 Preprint Update] Complex Systems Analysis of Arrested Neural Cell Differentiation during Development and Analogous Cell Cycling Models in Carcinogenesis V. I. Prisecaru and I.C. Baianu AFC-NMR & NIR Microspectroscopy Facility, College of ACES, FSHN & NPRE Departments, University of Illinois at Urbana, Urbana, IL. 61801, USA Email address: ibaianu @illinois.edu 1.Introduction A new approach to the modular, complex systems analysis of nonlinear dynamics of arrested neural cell Differentiation--induced cell proliferation during organismic development and the analogous cell cycling network transformations involved in carcinogenesis is proposed. Neural tissue arrested differentiation that induces cell proliferation during perturbed development and Carcinogenesis are complex processes that involve dynamically inter-connected biomolecules in the intercellular, membrane, cytosolic, nuclear and nucleolar compartments. Such 'dynamically inter-connected' biomolecules form numerous inter- related pathways referred to as 'molecular networks'. One such family of signaling pathways contains Nature Precedings : hdl:10101/npre.2012.7101.2 Posted 2 Apr 2012 the cell cyclins. Cyclins are proteins that link several critical pro-apoptotic and other cell cycling/division components, including the tumor suppressor gene TP53 and its product, the Thomsen-Friedenreich antigen (T antigen), Rb, mdm2, c-Myc, p21, p27, Bax, Bad and Bcl-2, which play major roles in various neoplastic transformations of many tissues. 1. Arrested Cell Differentiation in Neural -

Systems Biology 1 Systems Biology
Systems biology 1 Systems biology Systems biology is a term used to describe a number of trends in bioscience research, and a movement which draws on those trends. Proponents describe systems biology as a biology-based inter-disciplinary study field that focuses on complex interactions in biological systems, claiming that it uses a new perspective (holism instead of reduction). Particularly from year 2000 onwards, the term is used widely in the biosciences, and in a variety of contexts. An often stated ambition of An attempted illustration of the systems approach to biology systems biology is the modeling and discovery of emergent properties, properties of a system whose theoretical description is only possible using techniques which fall under the remit of systems biology. These typically involve cell signaling networks, via long-range allostery[1]. Overview Systems biology can be considered from a number of different aspects: • As a field of study, particularly, the study of the interactions between the components of biological systems, and how these interactions give rise to the function and behavior of that system (for example, the enzymes and metabolites in a metabolic pathway).[2][3] • As a paradigm, usually defined in antithesis to the so-called reductionist paradigm (biological organisation), although fully consistent with the scientific method. The distinction between the two paradigms is referred to in these quotations: "The reductionist approach has successfully identified most of the components and many of the interactions but, unfortunately, offers no convincing concepts or methods to understand how system properties emerge...the pluralism of causes and effects in biological networks is better addressed by observing, through quantitative measures, multiple components simultaneously and by rigorous data integration with mathematical models" Sauer et al[4] "Systems biology...is about putting together rather than taking apart, integration rather than reduction. -

FOOTPRINTS of GENERAL SYSTEMS THEORY Aleksandar Malecic Faculty of Electronic Engineering University of Nis, Serbia Aleks.Maleci
FOOTPRINTS OF GENERAL SYSTEMS THEORY Aleksandar Malecic Faculty of Electronic Engineering University of Nis, Serbia [email protected] ABSTRACT In order to identify General Systems Theory (GST) or at least have a fuzzy idea of what it might look like, we shall look for its traces on different systems. We shall try to identify such an “animal” by its “footprints”. First we mention some natural and artificial systems relevant for our search, than note the work of other people within cybernetics and identification of systems, and after that we are focused on what unification of different systems approaches and scientific disciplines should take into account (and whether or not it is possible). Axioms and principles are mentioned as an illustration of how to look for GST*. A related but still separate section is about Len Troncale and linkage propositions. After them there is another overview of different kinds of systems (life, consciousness, and physics). The documentary film Dangerous Knowledge and ideas of its characters Georg Cantor, Ludwig Boltzmann, Kurt Gödel, and Alan Turing are analyzed through systems worldview. “Patterns all the way down” and similar ideas by different authors are elaborated and followed by a section on Daniel Dennett’s approach to real patterns. Two following sections are dedicated to Brian Josephson (a structural theory of everything) and Sunny Auyang. After that the author writes about cosmogony, archetypes, myths, and dogmas. The paper ends with a candidate for GST*. Keywords: General Systems Theory, unification, patterns, principles, linkage propositions SYSTEMS IN NATURE AND ENGINEERING The footprints manifest as phenomena and scientific disciplines. -

Minireview Challenges for Complex Microbial Ecosystems: Combination of Experimental Approaches with Mathematical Modeling
Microbes Environ. Vol. 28, No. 3, 285–294, 2013 https://www.jstage.jst.go.jp/browse/jsme2 doi:10.1264/jsme2.ME13034 Minireview Challenges for Complex Microbial Ecosystems: Combination of Experimental Approaches with Mathematical Modeling SHIN HARUTA1, TAKEHITO YOSHIDA2, YOSHITERU AOI3,4, KUNIHIKO KANEKO5, and HIROYUKI FUTAMATA6* 1Department of Biological Sciences, Graduate School of Science and Engineering, Tokyo Metropolitan University, 1–1 Minami-Osawa, Hachioji, Tokyo 192–0397, Japan; 2Department of General Systems Studies, University of Tokyo 3–8–1 Komaba, Meguro, Tokyo 153–8902, Japan; 3Institute for Sustainable Sciences and Development, Hiroshima University, 2–113 Kagamiyama, Higashi-Hiroshima, Hiroshima 739–8527, Japan; 4Department of Biology, Northeastern University, 360 Huntington Ave. Mugar Lifescience Bldg., Boston, MA 02115, USA; 5Research Center for Complex Systems Biology, University of Tokyo, Meguro-ku, Tokyo, 153–8902, Japan; and 6Department of Applied Chemistry and Biochemical Engineering, Graduate School of Engineering, Shizuoka University, Hamamatsu, 432–8561, Japan (Received March 12, 2013—Accepted June 7, 2013—Published online August 30, 2013) In the past couple of decades, molecular ecological techniques have been developed to elucidate microbial diversity and distribution in microbial ecosystems. Currently, modern techniques, represented by meta-omics and single cell observations, are revealing the incredible complexity of microbial ecosystems and the large degree of phenotypic variation. These studies propound that microbiological techniques are insufficient to untangle the complex microbial network. This minireview introduces the application of advanced mathematical approaches in combination with microbiological experiments to microbial ecological studies. These combinational approaches have successfully elucidated novel microbial behaviors that had not been recognized previously. Furthermore, the theoretical perspective also provides an understanding of the plasticity, robustness and stability of complex microbial ecosystems in nature. -

JUNE: Open-Source Individual-Based Epidemiology Simulation
medRxiv preprint doi: https://doi.org/10.1101/2020.12.15.20248246; this version posted December 16, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted medRxiv a license to display the preprint in perpetuity. It is made available under a CC-BY-NC-ND 4.0 International license . JUNE open-source individual-based epidemiology simulation Joseph Bullock1,2,*, Carolina Cuesta-Lazaro1,3,*, Arnau Quera-Bofarull1,3,*, Miguel Icaza-Lizaola1,3,**, Aidan Sedgewick1,4,**, Henry Truong1,2,**, Aoife Curran1,3, Edward Elliott1,3, Tristan Caulfield7, Kevin Fong8,9, Ian Vernon1,6, Julian Williams5, Richard Bower1,3, and Frank Krauss1,2,+ 1 Institute for Data Science, Durham University, Durham DH1 3LE, UK 2Institute for Particle Physics Phenomenology, Durham University, Durham DH1 3LE, UK 3Institute for Computational Cosmology, Durham University, Durham DH1 3LE, UK 4Centre for Extragalactic Astronomy, Durham University, Durham DH1 3LE, UK 5Institute for Hazard, Risk & Resilience, Durham University, Durham DH1 3LE, UK 6Department of Mathematical Sciences, Durham University, Durham DH1 3LE, UK 7Department of Computer Science, University College of London, London WC1E 6BT, UK 8Department of Science, Technology, Engineering and Public Policy, University College London, London WC1E 6BT, UK 9Department of Anaesthesia, University College London Hospital, London NW1 2BU, UK *Equal contribution **Equal contribution +Corresponding author: [email protected] Abstract: We introduce JUNE, an open-source framework for the detailed simulation of epi- demics on the basis of social interactions in a virtual population constructed from geographically granular census data, reflecting age, sex, ethnicity, and socio-economic indicators. -

Systems Biology Success Stories In
SUCCESS STORIES IN SYSTEMS BIOLOGY Project funded by the European Comission under the Seventh Framework Programme for Research and Technological Development Contents Welcome 2 Research 3 A model that gets to the heart of systems biology 3 Denis Noble, Oxford University Taking a systems-eye view of cancers in children 5 Walter Kolch, Systems Biology Ireland Systems biology study points to Turing model of finger formation 7 James Sharpe, Centre for Genomic Regulation Balancing flavour and texture in tomatoes - with systems biology 9 Stuart Dunbar, Syngenta Systems X.ch: Swiss bank on systems biology 11 Daniel Vonder Muehll, SystemsX, Gisou van der Goot, EPF Lausanne, Cris Kuhlemeier, University of Bern Answering big questions with a small bug 13 Luis Serrano, Centre for Genomic Regulation Blueprints for life 15 Bas Teusink, Netherlands Platform for Systems Biology Merrimack: Following a systems path to drug discovery 17 Peter Sorger, Massachusetts Institute of Technology, and Birgit Schoeberl, Merrimack Pharmaceuticals Beating the conundrum of variability in cardiac simulations 19 Blanca Rodriguez, Oxford University Stats and modelling – a Belgian diagnostic tool for arthritis 21 Thibault Helleputte, DNAlytics Virtual Liver Network: a collaborative solution to hepatic diseases 23 Adriano Henney, Virtual Liver Network Tools and Resources 25 SBML: A lingua franca for systems biology 25 Michael Hucka, California Institute of Technology Model Support: JWS Online and BioModels Database 27 Jacky Snoep, Stellenbosch University, and Henning Hermjakob, -

The Challenges Facing Systemic Approaches in Biology: an Interview with Kunihiko Kaneko
View metadata, citation and similar papers at core.ac.uk brought to you by CORE PERSPECTIVE ARTICLE published: 05 December 2011 provided by PubMed Central doi: 10.3389/fphys.2011.00093 The challenges facing systemic approaches in biology: an interview with Kunihiko Kaneko Kunihiko Kaneko* Research Center for Complex Systems Biology, University of Tokyo, Tokyo, Japan Edited by: Interviewed by Kumar Selvarajoo and MasaTsuchiya at Institute for Advanced Biosciences, Kumar Selvarajoo, Keio University, Keio University, Japan, I discuss my approach to biology, what I call complex systems biol- Japan ogy. The approach is constructive in nature, and is based on dynamical systems theory Reviewed by: Kumar Selvarajoo, Keio University, and statistical physics. It is intended to understand universal characteristics of life sys- Japan tems; generic adaptation under noise, differentiation from stem cells in interacting cells, Masa Tsuchiya, Keio University, Japan robustness and plasticity in evolution, and so forth. Current status and future directions in *Correspondence: systems biology in Japan are also discussed. Kunihiko Kaneko, Research Center for Complex Systems Biology, University Keywords: plasticity, adaptation, robustness, stem cell, noise of Tokyo, Meguro, Komaba, Tokyo 153-8902, Japan. e-mail: [email protected]. ac.jp Q1: PROFESSOR KANEKO, CAN YOU INTRODUCE YOURSELF for the Logic of Life as a Complex System” (1999–2004) and AND YOUR RESEARCH? the ERATO project ”Kaneko Complex Systems Biology” (2004– I have been a physicist, and although I am more and more involved 2010), and am a head of Center for Complex Systems Biology at in biology now, I think my approach is quite a physicist-type. -

Meeting New Challenges in Food Science Technology: the Development of Complex Systems Approach for Food and Biobased Research
Meeting new challenges in food science technology: the development of complex systems approach for food and biobased research Hugo de Vries, Monique Axelos, Pascale Sarni-Manchado, Michael O’Donohue To cite this version: Hugo de Vries, Monique Axelos, Pascale Sarni-Manchado, Michael O’Donohue. Meeting new chal- lenges in food science technology: the development of complex systems approach for food and biobased research. Innovative Food Science and Emerging Technologies, Elsevier, 2018, 46, pp.1-6. 10.1016/j.ifset.2018.04.004. hal-01837847 HAL Id: hal-01837847 https://hal.archives-ouvertes.fr/hal-01837847 Submitted on 26 May 2020 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Distributed under a Creative Commons Attribution| 4.0 International License Accepted Manuscript Meeting new challenges in food science technology: the development of complex systems approach for food and biobased research Hugo de Vries, Monique Axelos, Pascale Sarni-Manchado, Michael O'Donohue PII: S1466-8564(18)30422-3 DOI: doi:10.1016/j.ifset.2018.04.004 Reference: INNFOO 1965 To appear in: Innovative Food Science and Emerging Technologies Please cite this article as: Hugo de Vries, Monique Axelos, Pascale Sarni-Manchado, Michael O'Donohue , Meeting new challenges in food science technology: the development of complex systems approach for food and biobased research. -
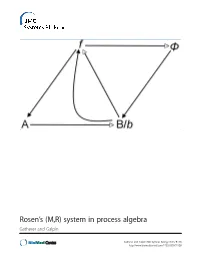
Rosen's (M,R) System in Process Algebra
Rosen’s (M,R) system in process algebra Gatherer and Galpin Gatherer and Galpin BMC Systems Biology 2013, 7:128 http://www.biomedcentral.com/1752-0509/7/128 Gatherer and Galpin BMC Systems Biology 2013, 7:128 http://www.biomedcentral.com/1752-0509/7/128 RESEARCH ARTICLE Open Access Rosen’s (M,R) system in process algebra Derek Gatherer1,3* and Vashti Galpin2 Abstract Background: Robert Rosen’s Metabolism-Replacement, or (M,R), system can be represented as a compact network structure with a single source and three products derived from that source in three consecutive reactions. (M,R) has been claimed to be non-reducible to its components and algorithmically non-computable, in the sense of not being evaluable as a function by a Turing machine. If (M,R)-like structures are present in real biological networks, this suggests that many biological networks will be non-computable, with implications for those branches of systems biology that rely on in silico modelling for predictive purposes. Results: We instantiate (M,R) using the process algebra Bio-PEPA, and discuss the extent to which our model represents a true realization of (M,R). We observe that under some starting conditions and parameter values, stable states can be achieved. Although formal demonstration of algorithmic computability remains elusive for (M,R), we discuss the extent to which our Bio-PEPA representation of (M,R) allows us to sidestep Rosen’s fundamental objections to computational systems biology. Conclusions: We argue that the behaviour of (M,R) in Bio-PEPA shows life-like properties. Keywords: Robert Rosen, (M,R), Metabolism-replacement, Metabolism-repair, Relational biology, Process algebra, Bio-PEPA, Computability, Turing machine Background kind. -

Complex Systems Theory and Biodynamics Complexity, Emergent Systems and Complex Biological Systems
Complex Systems Theory and Biodynamics Complexity, Emergent Systems and Complex Biological Systems PDF generated using the open source mwlib toolkit. See http://code.pediapress.com/ for more information. PDF generated at: Tue, 09 Jun 2009 07:58:48 UTC System 2 Complex Systems Theory System System (from Latin systēma, in turn from Greek σύστημα systēma) is a set of interacting or interdependent entities, real or abstract, forming an integrated whole. The concept of an 'integrated whole' can also be stated in terms of a system embodying a set of relationships which are differentiated from relationships of the set to other elements, and from relationships between an element of the set and elements not a part of the relational regime. The scientific research field which is engaged in the study of the general A schematic representation of a closed system and its properties of systems include systems boundary theory, systems science, systemics and systems engineering. They investigate the abstract properties of the matter and organization, searching concepts and principles which are independent of the specific domain, substance, type, or temporal scales of existence. Most systems share the same common characteristics. These common characteristics include the following • Systems are abstractions of reality. • Systems have structure which is defined by its parts and their composition. • Systems have behavior, which involves inputs, processing and outputs of material, information or energy. • Systems have interconnectivity, the various parts of a system have functional as well as structural relationships between each other. The term system may also refer to a set of rules that governs behavior or structure. -

A Framework for Evolutionary Systems Biology Laurence Loewe
BMC Systems Biology BioMed Central Correspondence Open Access A framework for evolutionary systems biology Laurence Loewe Address: Centre for Systems Biology at Edinburgh, The University of Edinburgh, Darwin building, Kings Buildings, Mayfield Road, Edinburgh, Scotland, EH9 3JU, UK Email: Laurence Loewe - [email protected] Published: 24 February 2009 Received: 1 July 2008 Accepted: 24 February 2009 BMC Systems Biology 2009, 3:27 doi:10.1186/1752-0509-3-27 This article is available from: http://www.biomedcentral.com/1752-0509/3/27 © 2009 Loewe; licensee BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Abstract Background: Many difficult problems in evolutionary genomics are related to mutations that have weak effects on fitness, as the consequences of mutations with large effects are often simple to predict. Current systems biology has accumulated much data on mutations with large effects and can predict the properties of knockout mutants in some systems. However experimental methods are too insensitive to observe small effects. Results: Here I propose a novel framework that brings together evolutionary theory and current systems biology approaches in order to quantify small effects of mutations and their epistatic interactions in silico. Central to this approach is the definition of fitness correlates that can be computed in some current systems biology models employing the rigorous algorithms that are at the core of much work in computational systems biology.