NCATS Bioplanet of Pathways
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Kyoto Encyclopedia of Genes and Genomes (KEGG)
Kyoto Encyclopedia of Genes and Genome Minoru Kanehisa Institute for Chemical Research, Kyoto University HFSPO Workshop, Strasbourg, November 18, 2016 The KEGG Databases Category Database Content PATHWAY KEGG pathway maps Systems information BRITE BRITE functional hierarchies MODULE KEGG modules KO (KEGG ORTHOLOGY) KO groups for functional orthologs Genomic information GENOME KEGG organisms, viruses and addendum GENES / SSDB Genes and proteins / sequence similarity COMPOUND Chemical compounds GLYCAN Glycans Chemical information REACTION / RCLASS Reactions / reaction classes ENZYME Enzyme nomenclature DISEASE Human diseases DRUG / DGROUP Drugs / drug groups Health information ENVIRON Health-related substances (KEGG MEDICUS) JAPIC Japanese drug labels DailyMed FDA drug labels 12 manually curated original DBs 3 DBs taken from outside sources and given original annotations (GENOME, GENES, ENZYME) 1 computationally generated DB (SSDB) 2 outside DBs (JAPIC, DailyMed) KEGG is widely used for functional interpretation and practical application of genome sequences and other high-throughput data KO PATHWAY GENOME BRITE DISEASE GENES MODULE DRUG Genome Molecular High-level Practical Metagenome functions functions applications Transcriptome etc. Metabolome Glycome etc. COMPOUND GLYCAN REACTION Funding Annual budget Period Funding source (USD) 1995-2010 Supported by 10+ grants from Ministry of Education, >2 M Japan Society for Promotion of Science (JSPS) and Japan Science and Technology Agency (JST) 2011-2013 Supported by National Bioscience Database Center 0.8 M (NBDC) of JST 2014-2016 Supported by NBDC 0.5 M 2017- ? 1995 KEGG website made freely available 1997 KEGG FTP site made freely available 2011 Plea to support KEGG KEGG FTP academic subscription introduced 1998 First commercial licensing Contingency Plan 1999 Pathway Solutions Inc. -

Precise Generation of Systems Biology Models from KEGG Pathways Clemens Wrzodek*,Finjabuchel,¨ Manuel Ruff, Andreas Drager¨ and Andreas Zell
Wrzodek et al. BMC Systems Biology 2013, 7:15 http://www.biomedcentral.com/1752-0509/7/15 METHODOLOGY ARTICLE OpenAccess Precise generation of systems biology models from KEGG pathways Clemens Wrzodek*,FinjaBuchel,¨ Manuel Ruff, Andreas Drager¨ and Andreas Zell Abstract Background: The KEGG PATHWAY database provides a plethora of pathways for a diversity of organisms. All pathway components are directly linked to other KEGG databases, such as KEGG COMPOUND or KEGG REACTION. Therefore, the pathways can be extended with an enormous amount of information and provide a foundation for initial structural modeling approaches. As a drawback, KGML-formatted KEGG pathways are primarily designed for visualization purposes and often omit important details for the sake of a clear arrangement of its entries. Thus, a direct conversion into systems biology models would produce incomplete and erroneous models. Results: Here, we present a precise method for processing and converting KEGG pathways into initial metabolic and signaling models encoded in the standardized community pathway formats SBML (Levels 2 and 3) and BioPAX (Levels 2 and 3). This method involves correcting invalid or incomplete KGML content, creating complete and valid stoichiometric reactions, translating relations to signaling models and augmenting the pathway content with various information, such as cross-references to Entrez Gene, OMIM, UniProt ChEBI, and many more. Finally, we compare several existing conversion tools for KEGG pathways and show that the conversion from KEGG to BioPAX does not involve a loss of information, whilst lossless translations to SBML can only be performed using SBML Level 3, including its recently proposed qualitative models and groups extension packages. -

Global LC/MS Metabolomics Profiling of Calcium Stressed and Immunosuppressant Drug Treated Saccharomyces Cerevisiae
Metabolites 2013, 3, 1102-1117; doi:10.3390/metabo3041102 OPEN ACCESS metabolites ISSN 2218-1989 www.mdpi.com/journal/metabolites/ Article Global LC/MS Metabolomics Profiling of Calcium Stressed and Immunosuppressant Drug Treated Saccharomyces cerevisiae Stefan Jenkins 1,2,3,*, Steven M. Fischer 2, Lily Chen 3 and Theodore R. Sana 2 1 Life Sciences Division, Lawrence Berkeley National Laboratory, Berkeley, CA 94720, USA 2 Agilent Technologies, Life Sciences, Diagnostics and Applied Markets, Santa Clara, CA 95051, USA; E-Mails: [email protected] (S.M.F.); [email protected] (T.R.S.) 3 Biology Department, San Francisco State University, San Francisco, CA 94132, USA; E-Mail: [email protected] * Author to whom correspondence should be addressed; E-Mail: [email protected]; Tel.: +1-650-922-5046; Fax: +1-510-486-4545. Received: 15 October 2013; in revised form: 20 November 2013 / Accepted: 25 November 2013 / Published: 6 December 2013 Abstract: Previous studies have shown that calcium stressed Saccharomyces cerevisiae, challenged with immunosuppressant drugs FK506 and Cyclosporin A, responds with comprehensive gene expression changes and attenuation of the generalized calcium stress response. Here, we describe a global metabolomics workflow for investigating the utility of tracking corresponding phenotypic changes. This was achieved by efficiently analyzing relative abundance differences between intracellular metabolite pools from wild-type and calcium stressed cultures, with and without prior immunosuppressant drugs exposure. We used pathway database content from WikiPathways and YeastCyc to facilitate the projection of our metabolomics profiling results onto biological pathways. A key challenge was to increase the coverage of the detected metabolites. This was achieved by applying both reverse phase (RP) and aqueous normal phase (ANP) chromatographic separations, as well as electrospray ionization (ESI) and atmospheric pressure chemical ionization (APCI) sources for detection in both ion polarities. -

And Macro-Level Network Metrics Unveils Top Communicative Gene Modules in Psoriasis
G C A T T A C G G C A T genes Article Using Micro- and Macro-Level Network Metrics Unveils Top Communicative Gene Modules in Psoriasis 1, 2 3 Reyhaneh Naderi y, Homa Saadati Mollaei , Arne Elofsson 3, , and Saman Hosseini Ashtiani * y 1 Department of Artificial Intelligence and Robotics, Faculty of Computer Engineering, Iran University of Science and Technology, Tehran 1684613114, Iran; [email protected] 2 Department of Advanced Sciences and Technology, Islamic Azad University Tehran Medical Sciences, Tehran 1916893813, Iran; [email protected] 3 Department of Biochemistry and Biophysics and Science for Life Laboratory, Stockholm University, 106 91 Stockholm, Sweden; [email protected] * Correspondence: [email protected]; Tel.: +46-762623644 These authors contributed equally to this work and should be considered joint first authors. y Received: 9 July 2020; Accepted: 6 August 2020; Published: 10 August 2020 Abstract: (1) Background: Psoriasis is a multifactorial chronic inflammatory disorder of the skin, with significant morbidity, characterized by hyperproliferation of the epidermis. Even though psoriasis’ etiology is not fully understood, it is believed to be multifactorial, with numerous key components. (2) Methods: In order to cast light on the complex molecular interactions in psoriasis vulgaris at both protein–protein interactions and transcriptomics levels, we studied a set of microarray gene expression analyses consisting of 170 paired lesional and non-lesional samples. Afterwards, a network analysis was conducted on the protein–protein interaction network of differentially expressed genes based on micro- and macro-level network metrics at a systemic level standpoint. (3) Results: We found 17 top communicative genes, all of which were experimentally proven to be pivotal in psoriasis, which were identified in two modules, namely the cell cycle and immune system. -

Biological Pathways
Biological pathways Bing Zhang Department of Biomedical Informatics Vanderbilt University [email protected] Biological pathway A biological pathway is a series of actions among molecules in a cell that leads to a certain product or a change in a cell. Different types of biological pathways Signal transduction pathways Gene regulation pathways Metabolic pathways 2 Applied Bioinformatics, Spring 2011 Outline Pathway databases Pathway assembly and editing Pathway mapping and enrichment analysis 3 Applied Bioinformatics, Spring 2011 Selected pathway databases KEGG Kyoto Encyclopedia of Genes and Genomes http://www.genome.jp/kegg/pathway.html Reactome Ontario Institute for Cancer Research, Cold Spring Harbor Laboratory, New York University School of Medicine and The European Bioinformatics Institute http://www.reactome.org/ WikiPathways University of Maastricht and UCSF http://www.wikipathways.org/ 4 Applied Bioinformatics, Spring 2011 KEGG: data model Molecular building blocks KEGG GENES: genes and proteins generated by genome sequencing projects KEGG ORTHOLOGY: orthology (KO) groups KEGG COMPOUND: small molecules KEGG REACTIONS: biochemical reactions KEGG PATHWAY: pathway maps Created in a general way to be applicable to all organisms, in terms of the orthologs defined by KO groups Organism-specific pathways can be generated by converting KO groups to gene identifiers in a given organism 5 Applied Bioinformatics, Spring 2011 KEGG: TCA cycle 6 Applied Bioinformatics, Spring 2011 Reactome: data model Entities -

Confirmation of Pathogenic Mechanisms by SARS-Cov-2–Host
Messina et al. Cell Death and Disease (2021) 12:788 https://doi.org/10.1038/s41419-021-03881-8 Cell Death & Disease ARTICLE Open Access Looking for pathways related to COVID-19: confirmation of pathogenic mechanisms by SARS-CoV-2–host interactome Francesco Messina 1, Emanuela Giombini1, Chiara Montaldo1, Ashish Arunkumar Sharma2, Antonio Zoccoli3, Rafick-Pierre Sekaly2, Franco Locatelli4, Alimuddin Zumla5, Markus Maeurer6,7, Maria R. Capobianchi1, Francesco Nicola Lauria1 and Giuseppe Ippolito 1 Abstract In the last months, many studies have clearly described several mechanisms of SARS-CoV-2 infection at cell and tissue level, but the mechanisms of interaction between host and SARS-CoV-2, determining the grade of COVID-19 severity, are still unknown. We provide a network analysis on protein–protein interactions (PPI) between viral and host proteins to better identify host biological responses, induced by both whole proteome of SARS-CoV-2 and specific viral proteins. A host-virus interactome was inferred, applying an explorative algorithm (Random Walk with Restart, RWR) triggered by 28 proteins of SARS-CoV-2. The analysis of PPI allowed to estimate the distribution of SARS-CoV-2 proteins in the host cell. Interactome built around one single viral protein allowed to define a different response, underlining as ORF8 and ORF3a modulated cardiovascular diseases and pro-inflammatory pathways, respectively. Finally, the network-based approach highlighted a possible direct action of ORF3a and NS7b to enhancing Bradykinin Storm. This network-based representation of SARS-CoV-2 infection could be a framework for pathogenic evaluation of specific 1234567890():,; 1234567890():,; 1234567890():,; 1234567890():,; clinical outcomes. -
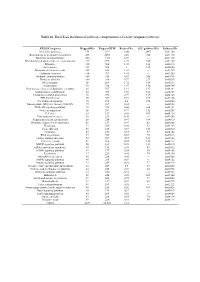
Table 1S. the KEGG Biochemical Pathways Categorization of LA Lily Unigenes Pathways
Table 1S. The KEGG biochemical pathways categorization of LA lily unigenes pathways. KEGG Categories Mapped-KO Unigene-NUM Ratio of No. ALL pathway KO Pathway-ID Metabolic pathways 954 4337 8.66 2067 ko01100 Biosynthesis of secondary metabolites 403 2285 4.56 720 ko01110 Biosynthesis of antibiotics 206 1147 2.29 --- ko01130 Microbial metabolism in diverse environments 157 995 1.99 720 ko01120 Ribosome 122 504 1.01 142 ko03010 Spliceosome 107 502 1 115 ko03040 Biosynthesis of amino acids 105 614 1.23 --- ko01230 Carbon metabolism 104 707 1.41 --- ko01200 Oxidative phosphorylation 100 335 0.67 206 ko00190 Purine metabolism 100 386 0.77 237 ko00230 RNA transport 98 861 1.72 134 ko03013 Endocytosis 92 736 1.47 138 ko04144 Protein processing in endoplasmic reticulum 88 567 1.13 137 ko04141 homologous recombination 84 933 1.86 144 ko05169 Ubiquitin mediated proteolysis 78 396 0.79 119 ko04120 HTLV-I infection 76 367 0.73 199 ko05166 Pyrimidine metabolism 76 298 0.6 150 ko00240 Non-alcoholic fatty liver disease (NAFLD) 72 209 0.42 --- ko04932 PI3K-Akt signaling pathway 71 328 0.66 226 ko04151 Viral carcinogenesis 68 387 0.77 132 ko05203 Cell cycle 63 339 0.68 103 ko04110 Proteoglycans in cancer 58 229 0.46 --- ko05205 Regulation of actin cytoskeleton 58 234 0.47 144 ko04810 Ribosome biogenesis in eukaryotes 56 237 0.47 82 ko03008 Phagosome 54 280 0.56 93 ko04145 Focal adhesion 53 185 0.37 133 ko04510 Lysosome 53 253 0.51 99 ko04142 RNA degradation 53 305 0.61 70 ko03018 Herpes simplex infection 52 257 0.51 121 ko05168 Cell cycle - yeast 51 260 -

Post-Graduate Diploma Program in Personalized Genomic Medicine” for Skill Development (DBT Sponsored Scheme)
UNIVERSITY OF MYSORE Syllabus for “Post-graduate Diploma Program in Personalized Genomic Medicine” For Skill Development (DBT Sponsored Scheme) FCBCS-CAGP SYSTEM Department of Studies in Genetics and Genomics University of Mysore Manasagangotri, Mysuru – 570 006 2019-20 1 PREAMBLE: The Information and potential use of genomic discoveries are no longer issues left for scientists and medical professionals to handle, but have become ones for the public at large. Rarely a day passes without genomics related story reported in the media. The proposed diploma program is designed to provide advanced knowledge dissemination in the field of genome sciences, applications and laboratory skills needed for molecular diagnostics and precision medicine procedures conducted in a clinical or research environment. The program is intended for those individuals who wish to enhance their laboratory expertise and knowledge in molecular-based methods. Precision medicine has the potential to fundamentally change how health care is practiced, but requires a trained health care workforce that understands the complexities of this field. One important component of precision medicine is the use of an individual’s genomic information to offer targeted treatment, tailored to the individual. Our course aims to provide participants with advanced knowledge of genomics, an overview of the clinical applications of genomic medicine, the skills to evaluate the clinical validity and utility of new tests, and an appreciation of the associated ethical and social issues inherent in this field. The course is geared towards individuals with a background in the biological sciences and a basic understanding of genetics. It is designed to be succinct and clinically focused, offering both conceptual and practical information about real-world applications of genomics. -

Signaling in the Innate Immune Response
Signaling in the innate immune response The MIT Faculty has made this article openly available. Please share how this access benefits you. Your story matters. Citation Kim, Dennis H. “Signaling in the Innate Immune Response.” WormBook (December 2015): 1–51 © 2015 WormBook As Published http://dx.doi.org/10.1895/wormbook.1.83.2 Publisher WormBook Version Final published version Citable link http://hdl.handle.net/1721.1/109292 Terms of Use Creative Commons Attribution Detailed Terms http://creativecommons.org/licenses/by/2.5/ Signaling in the innate immune response Dennis H. Kim1§ and Jonathan J. Ewbank2§ 1Department of Biology, Massachusetts Institute of Technology, Cambridge, Massachusetts, USA 2 Centre d’Immunologie de Marseille-Luminy, Aix Marseille Université UM2, INSERM, U1104, CNRS UMR7280, 13288 Marseille, France. Edited by Iva Greenwald Last revised September 16, 2015 §To whom correspondence should be addressed. E-mail: [email protected] or [email protected] WormBook Early Online, published on December 22, 2015 as doi: 10.1895/wormbook.1.83.2. 1 1. Introduction The maintenance of cellular and organismal homeostasis in the face of changes in the environment is essential for survival. Pathogenic microorganisms represent one type of environmental challenge. Plants and animals have developed immune systems to counter the threat of infection. The innate immune system is an ancient system of host defense against microbial pathogens present across a very broad range of species. Genetic studies of innate immunity in Drosophila and mammals have revealed an evolutionary conservation of signaling mechanisms (Medzhitov and Janeway, 1998), which has been corroborated in part by the study of innate immunity in Caenorhabditis elegans. -

A Tool for Retrieving Pathway Genes from KEGG Pathway Database
bioRxiv preprint doi: https://doi.org/10.1101/416131; this version posted September 13, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. KPGminer: A tool for retrieving pathway genes from KEGG pathway database A. K. M. Azad School of Biotechnology and Biomolecular Sciences, University of NSW, Chancellery Walk, Kensington, 2033, Australia Abstract Pathway analysis is a very important aspect in computational systems bi- ology as it serves as a crucial component in many computational pipelines. KEGG is one of the prominent databases that host pathway information associated with various organisms. In any pathway analysis pipelines, it is also important to collect and organize the pathway constituent genes for which a tool to automatically retrieve that would be a useful one to the practitioners. In this article, I present KPGminer, a tool that retrieves the constituent genes in KEGG pathways for various organisms and or- ganizes that information suitable for many downstream pathway analysis pipelines. We exploited several KEGG web services using REST APIs, par- ticularly GET and LIST methods to request for the information retrieval which is available for developers. Moreover, KPGminer can operate both for a particular pathway (single mode) or multiple pathways (batch mode). Next, we designed a crawler to extract necessary information from the re- sponse and generated outputs accordingly. KPGminer brings several key features including organism-specific and pathway-specific extraction of path- way genes from KEGG and always up-to-date information. -

TERRA Regulate the Transcriptional Landscape of Pluripotent Cells
RESEARCH ARTICLE TERRA regulate the transcriptional landscape of pluripotent cells through TRF1-dependent recruitment of PRC2 Rosa Marı´aMario´ n1†, Juan J Montero1†, Isabel Lo´ pez de Silanes1, Osvaldo Gran˜ a-Castro2, Paula Martı´nez1, Stefan Schoeftner1‡§, Jose´ Alejandro Palacios-Fa´ brega1#, Maria A Blasco1* 1Telomeres and Telomerase Group, Molecular Oncology Program, Spanish National Cancer Centre (CNIO), Madrid, Spain; 2Bioinformatics Unit, Structural Biology Program, Spanish National Cancer Centre (CNIO), Madrid, Spain Abstract The mechanisms that regulate pluripotency are still largely unknown. Here, we show that Telomere Repeat Binding Factor 1 (TRF1), a component of the shelterin complex, regulates the genome-wide binding of polycomb and polycomb H3K27me3 repressive marks to pluripotency genes, thereby exerting vast epigenetic changes that contribute to the maintenance of mouse ES *For correspondence: cells in a naı¨ve state. We further show that TRF1 mediates these effects by regulating TERRA, the [email protected] lncRNAs transcribed from telomeres. We find that TERRAs are enriched at polycomb and stem cell † These authors contributed genes in pluripotent cells and that TRF1 abrogation results in increased TERRA levels and in higher equally to this work TERRA binding to those genes, coincidental with the induction of cell-fate programs and the loss of Present address: ‡Genomic the naı¨ve state. These results are consistent with a model in which TRF1-dependent changes in Stability Unit, Laboratorio TERRA levels modulate polycomb -

Downregulated Developmental Processes in the Postnatal Right
www.nature.com/cddiscovery ARTICLE OPEN Downregulated developmental processes in the postnatal right ventricle under the influence of a volume overload ✉ ✉ ✉ Chunxia Zhou1,6, Sijuan Sun2,6, Mengyu Hu3, Yingying Xiao1, Xiafeng Yu1 , Lincai Ye 1,4,5 and Lisheng Qiu 1 © The Author(s) 2021 The molecular atlas of postnatal mouse ventricular development has been made available and cardiac regeneration is documented to be a downregulated process. The right ventricle (RV) differs from the left ventricle. How volume overload (VO), a common pathologic state in children with congenital heart disease, affects the downregulated processes of the RV is currently unclear. We created a fistula between the abdominal aorta and inferior vena cava on postnatal day 7 (P7) using a mouse model to induce a prepubertal RV VO. RNAseq analysis of RV (from postnatal day 14 to 21) demonstrated that angiogenesis was the most enriched gene ontology (GO) term in both the sham and VO groups. Regulation of the mitotic cell cycle was the second-most enriched GO term in the VO group but it was not in the list of enriched GO terms in the sham group. In addition, the number of Ki67-positive cardiomyocytes increased approximately 20-fold in the VO group compared to the sham group. The intensity of the vascular endothelial cells also changed dramatically over time in both groups. The Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis of the downregulated transcriptome revealed that the peroxisome proliferators-activated receptor (PPAR) signaling pathway was replaced by the cell cycle in the top-20 enriched KEGG terms because of the VO.