Efficient Elimination of Primary B-ALL Cells in Vitro and in Vivo Using A
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

It Takes Two to Tango
HIGHLIGHTS IMMUNOTHERAPY It takes two to tango Much of the recent work on the development of active, specific immunotherapy of cancer has focused on the induction of CD8+ cytotoxic T-lymphocyte (CTL) responses, and several studies have shown that it is possible to stimulate tumour-specific CTLs using dendritic cells (DCs) that are loaded with tumour antigens. But CD4+ T-helper type 1 (TH1) cells are also important day of vaccination and loaded with various So, the results from this Phase I trial provide components of effective immune responses. MHC class-I- and -II-restricted peptides. convincing evidence that cryopreserved DCs In this study, Schuler–Thurner and Patients with incurable melanoma were can induce TH1 responses against tumour colleagues provide the first evidence that DCs treated with five biweekly vaccinations of antigens without significant toxicity, and it that are loaded with tumour-specific peptides DCs, followed by assessment one month also encourages further development of can rapidly induce TH1 responses in cancer after the final vaccination. DC-based vaccination technology. patients that are readily detectable ex vivo. The results showed that the vaccination Elaine Bell References and links The DCs that were used for vaccination protocol induced a rapid TH1 response in in this study were derived from blood patients, both to a control immunizing antigen ORIGINAL RESEARCH PAPER Schuler–Thurner, B. et al. Rapid induction of tumor-specific type 1 T helper cells in monocytes that were matured ex vivo using and also to defined MHC class-II-restricted metastatic melanoma patients by vaccination with mature, a defined cocktail (consisting of interleukin tumour antigens. -

1 ICAM3-Fc Outperforms Receptor-Specific Antibodies
Preprints (www.preprints.org) | NOT PEER-REVIEWED | Posted: 10 April 2019 doi:10.20944/preprints201904.0118.v1 Peer-reviewed version available at Molecules 2019, 24, 1825; doi:10.3390/molecules24091825 ICAM3-Fc outperforms receptor-specific antibodies targeted nanoparticles to dendritic cells for cross-presentation Luis J. Cruz,1* Paul J. Tacken,2 Johan S. van der Schoot,2 Felix Rueda,3 Ruurd Torensma,2 Carl G. Figdor2* 1Translational Nanobiomaterials and Imaging, Department of Radiology, Leiden University Medical Center, Albinusdreef 2, 2333 ZA Leiden, The Netherlands. 2Department of Tumor Immunology, Radboud Insititute for Molecular Life Sciences, Radboud University Medical Center, Postbox 9101, 6500 HB Nijmegen, The Netherlands. 3Department of Biochemistry and Molecular Biology, University of Barcelona, Diagonal 643, 08028 Barcelona, Spain. Running title: ICAM3-Fc- versus antibody-targeted NP vaccines to human DCs Keywords: Delivery system; nanoparticle; targeting. AUTHOR INFORMATION Corresponding Author *Dr. Luis J. Cruz Translational Nanobiomaterials and Imaging, Department of Radiology, Bldg.1, C5-60. Leiden University Medical Center, Albinusdreef 2 2333 ZA Leiden, The Netherlands Tel: +31 71 5263025 Email: [email protected] *Prof. Dr. Carl G. Figdor Department of Tumor Immunology, Nijmegen Centre for Molecular Life Sciences, Radboud University Nijmegen Medical Centre, Postbox 9101, 6500 HB Nijmegen, The Netherlands. Fax: +31-24-3540339; Tel.: +31-24-3617600; E-mail: [email protected] 1 © 2019 by the author(s). Distributed -

Regulation of Calreticulin–Major Histocompatibility Complex (MHC) Class I Interactions by ATP
Regulation of calreticulin–major histocompatibility complex (MHC) class I interactions by ATP Sanjeeva Joseph Wijeyesakerea, Jessica K. Gagnonb, Karunesh Arorab, Charles L. Brooks IIIb, and Malini Raghavana,1 aDepartment of Microbiology and Immunology, University of Michigan Medical School, Ann Arbor, MI 48109; and bDepartment of Chemistry, University of Michigan, Ann Arbor, MI 48109 Edited by Peter Cresswell, Yale University School of Medicine, New Haven, CT, and approved September 1, 2015 (received for review May 26, 2015) The MHC class I peptide loading complex (PLC) facilitates the assembly computational methods validated by experimental approaches, we of MHC class I molecules with peptides, but factors that regulate the identify residues within the globular domain of calreticulin that stability and dynamics of the assembly complex are largely unchar- are important for ATP binding and ATPase activity. Based on acterized. Based on initial findings that ATP, in addition to MHC class further investigations into the functional effects of calreticulin I-specific peptide, is able to induce MHC class I dissociation from the mutants with deficiencies in ATP interactions, we elucidate a key PLC, we investigated the interaction of ATP with the chaperone role for ATP in the regulation of PLC dynamics and the in- calreticulin, an endoplasmic reticulum (ER) luminal, calcium-bind- teraction of calreticulin with other cellular proteins. ing component of the PLC that is known to bind ATP. We combined computational and experimental measurements to identify residues Results within the globular domain of calreticulin, in proximity to the high- ATP Destabilizes the PLC, Promoting MHC Class I Release. Calreticulin- affinity calcium-binding site, that are important for high-affinity ATP deficient mouse embryonic fibroblasts (MEFs) reconstituted binding and for ATPase activity. -

Cell Biology from an Immune Perspective in This Lecture We Will
Harvard-MIT Division of Health Sciences and Technology HST.176: Cellular and Molecular Immunology Course Director: Dr. Shiv Pillai Cell Biology from an Immune Perspective In this lecture we will very briefly review some aspects of cell biology which are required as background knowledge in order to understand how the immune system works. These will include: 1. A brief overview of protein trafficking 2. Signal transduction 3. The cell cycle Some of these issues will be treated in greater depth in later lectures. Protein Trafficking/The Secretory Pathway: From an immune perspective the secretory compartment and structures enclosed by vesicles are “seen” in different ways from proteins that reside in the cytosol or the nucleus. We will briefly review the secretory and endocytic pathways and discuss the biogenesis of membrane proteins. Some of the issues that will be discussed are summarized in Figures 1-3. Early endosomes Late endosomes Lysosomes Golgi Vesiculo-Tubular Compartment ER Figure 1. An overview of the secretory pathway Early endosomes Late endosomes Multivesicular and multilamellar bodies Golgi ER Proteasomes Figure 2. Protein degradation occurs mainly in lysosomes and proteasomes Proteins that enter the cell from the environment are primarily degraded in lysosomes. Most cytosolic and nuclear proteins are degraded in organelles called proteasomes. Intriguingly these two sites of degradation are each functionally linked to distinct antigen presentation pathways, different kinds of MHC molecules and the activation of different categories of T cells. Integral membrane proteins maybe inserted into the membrane in a number of ways, the two most common of these ways being considered in Figure 3. -

Innate Immunity: MHC Class I As a Negative Regulator of TLR Signalling
RESEARCH HIGHLIGHTS IN BRIEF T RANSPLANTATION Handling complement for transplant success Haematopoietic cell transplantation (HCT) is used to treat cancer and blood disorders, but a potentially lethal complication is the development of graft-versus-host disease (GVHD). Total-body irradiation (TBI) is required to facilitate HCT and can promote GVHD by activating host dendritic cells (DCs), although the mechanisms involved are not completely understood. This study shows that, in mice, TBI causes host DCs to upregulate the complement components C3, C5a, factor B and factor D, and to downregulate CD55, a negative regulator of the complement cascade. In HCT using CD55-deficient donor T cells, recipient mice developed exacerbated GVHD, suggesting that upregulation of complement components by host DCs promotes the activation of allogeneic donor T cells. Indeed, less severe GVHD was seen in HCT using donor T cells that were deficient in the C3a and C5a receptors. Treatment of recipients with a C5a receptor antagonist during the post-transplantation period reduced GVHD development, and could be a useful strategy for preventing GVHD in humans after transplantation. ORIGINAL RESEARCH PAPER Kwan, W.-H. et al. Antigen-presenting cell-derived complement modulates graft-versus-host disease. J. Clin. Invest. 15 May 2012 (doi:10.1172/JCI61019) T CELLS ‘Leaky’ cytokine secretion by immunological synapses This study used live-cell imaging to investigate whether cytokine secretion by activated T cells is restricted by the immunological synapse to antigen-specific target cells or also affects bystander cells in the local environment. Responses to interferon-γ (IFNγ) were monitored by tracking the intracellular localization of a fluorescent STAT1 protein in ovalbumin- expressing and non-expressing astrocytes cultured with activated ovalbumin-specific CD8+ T cells. -

Cancer Immune Evasion Through Loss of MHC Class I Antigen Presentation
University of Massachusetts Medical School eScholarship@UMMS Open Access Publications by UMMS Authors 2021-03-09 Cancer Immune Evasion Through Loss of MHC Class I Antigen Presentation Karthik Dhatchinamoorthy University of Massachusetts Medical School Et al. Let us know how access to this document benefits ou.y Follow this and additional works at: https://escholarship.umassmed.edu/oapubs Part of the Amino Acids, Peptides, and Proteins Commons, Biological Factors Commons, Cancer Biology Commons, Hemic and Immune Systems Commons, Immunopathology Commons, Neoplasms Commons, and the Pathology Commons Repository Citation Dhatchinamoorthy K, Colbert JD, Rock KL. (2021). Cancer Immune Evasion Through Loss of MHC Class I Antigen Presentation. Open Access Publications by UMMS Authors. https://doi.org/10.3389/ fimmu.2021.636568. Retrieved from https://escholarship.umassmed.edu/oapubs/4636 Creative Commons License This work is licensed under a Creative Commons Attribution 4.0 License. This material is brought to you by eScholarship@UMMS. It has been accepted for inclusion in Open Access Publications by UMMS Authors by an authorized administrator of eScholarship@UMMS. For more information, please contact [email protected]. REVIEW published: 09 March 2021 doi: 10.3389/fimmu.2021.636568 Cancer Immune Evasion Through Loss of MHC Class I Antigen Presentation Karthik Dhatchinamoorthy, Jeff D. Colbert and Kenneth L. Rock* Department of Pathology, UMass Medical School, Worcester, MA, United States Major histocompatibility class I (MHC I) molecules bind peptides derived from a cell’s expressed genes and then transport and display this antigenic information on the cell surface. This allows CD8T cells to identify pathological cells that are synthesizing abnormal proteins, such as cancers that are expressing mutated proteins. -

The MHC Class I–Like Fc Receptor Promotes Humorally Mediated Autoimmune Disease Shreeram Akilesh, Stefka Petkova, Thomas J
Research article The MHC class I–like Fc receptor promotes humorally mediated autoimmune disease Shreeram Akilesh, Stefka Petkova, Thomas J. Sproule, Daniel J. Shaffer, Gregory J. Christianson, and Derry Roopenian The Jackson Laboratory, Bar Harbor, Maine, USA. The MHC class I family–like Fc receptor, FcRn, is normally responsible for extending the life span of serum IgG Ab’s, but whether this molecule contributes to autoimmune pathogenesis remains speculative. To deter- mine directly whether this function contributes to humoral autoimmune disease, we examined whether a defi- ciency in the FcRn heavy chain influences autoimmune arthritis in the K/BxN mouse model. FcRn deficiency conferred either partial or complete protection in the arthritogenic serum transfer and the more aggressive genetically determined K/BxN autoimmune arthritis models. The protective effects of an FcRn deficiency could be overridden with excessive amounts of pathogenic IgG Ab’s. The therapeutic saturation of FcRn by high-dose intravenous IgG (IVIg) also ameliorated arthritis, directly implicating FcRn blockade as a signifi- cant mechanism of IVIg’s anti-inflammatory action. The results suggest that FcRn is a potential therapeutic target that links the initiation and effector phases of humoral autoimmune disease. Introduction mice been used as a model for addressing this question with mixed At the inductive phase of a humoral autoimmune response, B results (refs. 9–16; D. Roopenian, unpublished observations). This cells, after encounter with APCs and T cells, undergo antigen- is not surprising because β2m controls many immunological and driven proliferation and differentiation into Ab-secreting plasma nonimmunological processes, including the development and cells. During the effector phase, Ab’s bind autoantigen leading function of CD8 T cells, natural T cells, conventional NK cells (17), to downstream events such as activation of complement, recruit- and iron homeostasis (18). -

Mechanisms of HIV Protein Degradation Into Epitopes: Implications for Vaccine Design
Viruses 2014, 6, 3271-3292; doi:10.3390/v6083271 OPEN ACCESS viruses ISSN 1999-4915 www.mdpi.com/journal/viruses Review Mechanisms of HIV Protein Degradation into Epitopes: Implications for Vaccine Design Marijana Rucevic †, Julie Boucau †, Jens Dinter †, Georgio Kourjian † and Sylvie Le Gall * Ragon Institute of MGH, MIT and Harvard, Massachusetts General Hospital and Harvard Medical School, Cambridge, MA 02139, USA; E-Mails: [email protected] (M.R.); [email protected] (J.B.); [email protected] (J.D.); [email protected] (G.K.) † These authors contributed equally to this work. * Author to whom correspondence should be addressed; E-Mail: [email protected]; Tel.: +1-857-268-7010. Received: 11 June 2014; in revised form: 6 August 2014 / Accepted: 11 August 2014 / Published: 21 August 2014 Abstract: The degradation of HIV-derived proteins into epitopes displayed by MHC-I or MHC-II are the first events leading to the priming of HIV-specific immune responses and to the recognition of infected cells. Despite a wealth of information about peptidases involved in protein degradation, our knowledge of epitope presentation during HIV infection remains limited. Here we review current data on HIV protein degradation linking epitope production and immunodominance, viral evolution and impaired epitope presentation. We propose that an in-depth understanding of HIV antigen processing and presentation in relevant primary cells could be exploited to identify signatures leading to efficient or inefficient epitope presentation in HIV proteomes, and to improve the design of immunogens eliciting immune responses efficiently recognizing all infected cells. Keywords: HIV; antigen processing; protein degradation; proteasome; aminopeptidase; peptidase; immunogen; vaccine vector; dendritic cells; T cells; viral evolution Viruses 2014, 6 3272 1. -

Differential Colon Cancer Cell Adhesion to E-, P-, and L-Selectin: Role of Mucin- Type Glycoproteins1
[CANCER RESEARCH 55. 4425-4431, October 1, 1995] Differential Colon Cancer Cell Adhesion to E-, P-, and L-selectin: Role of Mucin- type Glycoproteins1 Gianna Mannori,2 Pascal Crottet,3 Oliviero Cecconi, Kohji Hanasaki, Alejandro Aruffo, Richard M. Nelson, Ajit Varki, and Michael P. Bevilacqua4 Howard Hughes Medical Institute, Cellular and Molecular Medicine and Department of Pathology, University of California at San Diego, La lolla, California 92093-0669 ¡C.M., O. C., K. H., R. M. N., M. P. B.]; Cancer Center, Glycobiology Program, University of California at San Diego, La Jolla, California 92093-0063 [P. C., A. VJ; and Bristol-Myers Squibb Pharmaceutical Research Institute, Seattle. Washington 98121 ¡A.A.J ABSTRACT trophils, monocytes, and most lymphocytes constitutively express L-selectin, a molecule that supports adhesive interactions with high E-, P-, and L-selectin support the adhesion of leukocytes to the vessel endothelial venules of lymph nodes, as well as activated endothelium wall through the recognition of specific carbohydrate ligands, which often contain sialylated, fucosylated lactosamines such as sialyl Lewis x [sLe"; at sites of inflammation (reviewed in Refs. 10-13). Several recent Neu5Aca2-3Gal/31-4(Fucal-3)GlcNAc-]. E-selectin expressed by acti studies have demonstrated that certain tumor cells interact with se vated endothelium has been shown to support the adhesion of sLex- lectins. In particular, E-selectin has been shown to mediate adhesion bearing colon cancer cells. In the present study, we examine the interac of colon cancer cells to activated vascular endothelium (14-21). In tions of multiple colon cancer cell lines with all three selectins. -
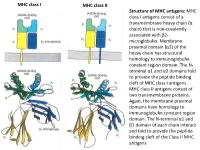
MHC Class I Antigens Consist of a Transmembrane Heavy Chain (Α Chain) That Is Non-Covalently Associated with Β2- Microglobulin
MHC class I MHC class II Structure of MHC antigens: MHC class I antigens consist of a transmembrane heavy chain (α chain) that is non-covalently associated with β2- microglobulin. Membrane proximal domain (α3) of the heavy chain has structural homology to immunoglobulin constant region domain. The N- terminal α1 and α2 domains fold to provide the peptide binding cleft of MHC class I antigens. MHC class II antigens consist of two transmembrane proteins. Again, the membrane proximal domains have homology to immunoglobulin constant region domain. The N-terminal α1 and β1 domain of each chain interact and fold to provide the peptide binding cleft of the Class II MHC antigens MHC class I MHC class II A top-down view of the peptide binding cleft of MHC antigens. The peptide binding cleft for MHC class I antigens is relatively narrower at the ends and accommodate 8- 10 amino acid residues long peptides. In contrast, due to non-covalent association between the N-terminal domains of the two chains of the class II antigens, the peptide binding cleft is open at the ends. This permits binding of larger peptides, typically 13-20 aa long. T cell receptor Interaction between T cell receptor and MHC-peptide complex: The T cell receptors binds to the peptide-MHC complex in a diagonal fashion. The CDR1 and CDR2 regions of the α and β chains of the T cell receptor bind to the α-helical walls of the peptide binding cleft. The CDR3 regions on the other hand bind to the peptide present in the cleft. -

The Meaning of Self
THE MEANING OF SELF MAJOR HISTOCOMPATIBILITY COMPLEX antigens and their role in the immune system Role of the IMMUNE SYSTEM: Hostility towards others Elimination of pathogens (bacterial and viral infections) Elimination of toxins and poisons Elimination of infected and malfunctioning cells Elimination of malignantly transformed cells with self-regard Preservation of healthy self-tissues intact MUST BE ABLE TO DISCRIMINATE SELF FROM NONSELF Subject: Re: the meaning of self From: Alan Challoner MA(Phil.) MChS 1. One of the most important aspects of personal development is the way in which we see ourselves. 2. As a child grows, he becomes aware through his experiences, initially within the family and later also outside in his society, of who and what he is. 3. This reality not only represents his present situation but also acts as a stepping-stone towards his future development. 4. This of course includes some degree of self-regard, for as Horney, 1950 and Rogers, 1951 have indicated, unless an individual loves himself, he will feel a basic hostility towards others. (random citation from the internet) MHC antigens (class I and class II) coordinate nearly all pathways of SELF –NONSELF discrimination in the adaptive immune system 1. During early development MHC antigens educate immune cells how to distinguish foe from friend based on what is present in the host environment 2. This “education” will have lasting effects on how the host will react to “self” and “nonself” in the future 3. MHC instruct immune system to be hostile to pathogens 4. MHC teach immune system to be tolerant to healthy tissues of the host MHC class I proteins act as molecular organizers of “self” and “nonself” peptides. -

HIV-1) CD4 Receptor and Its Central Role in Promotion of HIV-1 Infection
MICROBIOLOGICAL REVIEWS, Mar. 1995, p. 63–93 Vol. 59, No. 1 0146-0749/95/$04.0010 Copyright q 1995, American Society for Microbiology The Human Immunodeficiency Virus Type 1 (HIV-1) CD4 Receptor and Its Central Role in Promotion of HIV-1 Infection STEPHANE BOUR,* ROMAS GELEZIUNAS,† AND MARK A. WAINBERG* McGill AIDS Centre, Lady Davis Institute-Jewish General Hospital, and Departments of Microbiology and Medicine, McGill University, Montreal, Quebec, Canada H3T 1E2 INTRODUCTION .........................................................................................................................................................63 RETROVIRAL RECEPTORS .....................................................................................................................................64 Receptors for Animal Retroviruses ........................................................................................................................64 CD4 Is the Major Receptor for HIV-1 Infection..................................................................................................65 ROLE OF THE CD4 CORECEPTOR IN T-CELL ACTIVATION........................................................................65 Structural Features of the CD4 Coreceptor..........................................................................................................65 Interactions of CD4 with Class II MHC Determinants ......................................................................................66 CD4–T-Cell Receptor Interactions during T-Cell Activation