A High-Quality Genome of the Mangrove Aegiceras Corniculatum Aids Investigation of Molecular Adaptation to Intertidal Environments
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Incorporating the Plant Phenological Trajectory Into Mangrove Species Mapping with Dense Time Series Sentinel-2 Imagery and the Google Earth Engine Platform
remote sensing Article Incorporating the Plant Phenological Trajectory into Mangrove Species Mapping with Dense Time Series Sentinel-2 Imagery and the Google Earth Engine Platform Huiying Li 1,2, Mingming Jia 1,3,4,* , Rong Zhang 1, Yongxing Ren 1,5 and Xin Wen 1,5 1 Key Laboratory of Wetland Ecology and Environment, Northeast Institute of Geography and Agroecology, Chinese Academy of Sciences, Changchun 130102, China; [email protected] (H.L.); zrfi[email protected] (R.Z.); [email protected] (Y.R.); [email protected] (X.W.) 2 School of Management Engineering, Qingdao University of Technology, Qingdao 266520, China 3 State Key Laboratory of Information Engineering in Surveying, Mapping and Remote Sensing, Wuhan University, Wuhan 430079, China 4 National Earth System Science Data Center, Beijing 100101, China 5 College of Earth Sciences, Jilin University, Changchun 130061, China * Correspondence: [email protected] Received: 12 September 2019; Accepted: 22 October 2019; Published: 24 October 2019 Abstract: Information on mangrove species composition and distribution is key to studying functions of mangrove ecosystems and securing sustainable mangrove conservation. Even though remote sensing technology is developing rapidly currently, mapping mangrove forests at the species level based on freely accessible images is still a great challenge. This study built a Sentinel-2 normalized difference vegetation index (NDVI) time series (from 2017-01-01 to 2018-12-31) to represent phenological trajectories of mangrove species and then demonstrated the feasibility of phenology-based mangrove species classification using the random forest algorithm in the Google Earth Engine platform. It was found that (i) in Zhangjiang estuary, the phenological trajectories (NDVI time series) of different mangrove species have great differences; (ii) the overall accuracy and Kappa confidence of the classification map is 84% and 0.84, respectively; and (iii) Months in late winter and early spring play critical roles in mangrove species mapping. -

Short Communication Screening of Some Bangladeshi Medicinal Plants for in Vitro Antibacterial Activity
OPEM www.opem.org Oriental Pharmacy and Experimental Medicine 2008 8(3), 316-321 DOI 10.3742/OPEM.2008.8.3.316 Short Communication Screening of some Bangladeshi medicinal plants for in vitro antibacterial activity 1 1 1 1 2 Shaikh Jamal Uddin , Razina Rouf , Jamil Ahmed Shilpi , Mohammad Alamgir , Lutfun Nahar 2, and Satyajit Dey Sarker * 1 2 Pharmacy Discipline, Life Science School, Khulna University, Khulna-9208, Bangladesh; School of Biomedical Sciences, University of Ulster at Coleraine, Cromore Road, Coleraine BT52 1SA, Co. Londodnderry, Northern Ireland, UK Received for publication September 28, 2006; accepted April 08, 2008 SUMMARY A total of 33 extracts representing 26 plant species belonging to 24 families were collected from different regions of Bangladesh, and screened for their in vitro antibacterial activity against several pathogenic Gram-positive and Gram-negative bacterial strains using the conventional disc diffusion method. The most potent activity was exhibited by the extracts of Aegiceras corniculatum, Alocasia fornicata, Ceriops decandra, Cuscuta reflexa, Lasia spinosa, Lantana camara, Pandanus foetidus and Xylocarpus granatum. The extracts of Abtilon indicum, Derris trifoliata, Dendrophthoe falcat, Ruellia tuberosa and X. moluccensis did not show any antibacterial properties at test concentrations. Key words: Antibacterial activity; Medicinal plants; Disc diffusion method; Bangladesh INTRODUCTION significantly on the uses of medicinal plants. Bangladesh is one of the South Asian countries that Natural products have long been utilized as an have a rich and prestigious heritage of medicinal indispensable source for the discovery and plants’ uses to treat various diseases. As a development of new drugs. One of the major consequence, medicinal plants have traditionally sources of these natural products is medicinal occupied an important status in the socio-cultural, plants that have been used traditionally in many spiritual and medicinal arena of rural and tribal countries to combat various diseases, including lives on Bangladesh. -

Appendix 6.1 Plant Species Recorded in the Assessment Area
Appendix 6.1 Plant Species Recorded in the Assessment Area Assessment Area Area within Urbanised/ Site Relative Mixed Tall Low Abandoned disturbed Mangrove/ Other Formation Species Habit Exotic Abundance Woodland Plantation Shrubland Shrubland Field Area Sandflat Coastal Stream Boundary Abrus mollis C Scarce Y Acacia confusa T Exotic Common Y Y Y Y Y Acacia auriculiformis T Exotic Common Y Y Acacia mangium T Exotic Scarce Y Acanthus ilicifolius S Scarce Y Acorus gramineus H Occasional Y Acronychia pedunculata T Common Y Y Y Y Acrostichum aureum F Scarce Y Adiantum capillus-veneris F Scarce Y Y Adiantum flabellatum T Exotic Common Y Y Y Adinandra millettii T Common Y Y Y Y Aegiceras corniculatum S Scarce Y Y Y Agave angustifolia H Exotic Scarce Y Ageratum conyzoides H Common Y Y Y Aglaia odorata S Exotic Scarce Y Y Y Alangium chinense T Occasional Y Albizia lebbeck T Occasional Y Y Y Alocasia odora H Common Y Y Y Y Aloe vera H Exotic Scarce Y Alpinia zerumbet H Common Y Y Y Alpinia zerumbet cv. Variegata H Exotic Occasional Y Alyxia sinensis C Scarce Y Y Antidesma ghaesembilla S Scarce Y Y Antirhea chinensis T Scarce Y Aporosa dioica T Common Y Y Y Y Y Y Aquilaria sinensis T Occasional Y Y Y Y Aralia armata T Occasional Y Y Araucaria heterophylla T Exotic Scarce Y Y Archontophoenix alexandrae T Exotic Scarce Y Assessment Area Area within Urbanised/ Site Relative Mixed Tall Low Abandoned disturbed Mangrove/ Other Formation Species Habit Exotic Abundance Woodland Plantation Shrubland Shrubland Field Area Sandflat Coastal Stream Boundary Ardisia crenata S Occasional Y Y Y Ardisia quinquegona S Occasional Y Ardisia punctata S Scarce Y Artemisia sp. -

The 1770 Landscape of Botany Bay, the Plants Collected by Banks and Solander and Rehabilitation of Natural Vegetation at Kurnell
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Hochschulschriftenserver - Universität Frankfurt am Main Backdrop to encounter: the 1770 landscape of Botany Bay, the plants collected by Banks and Solander and rehabilitation of natural vegetation at Kurnell Doug Benson1 and Georgina Eldershaw2 1Botanic Gardens Trust, Mrs Macquaries Rd Sydney 2000 AUSTRALIA email [email protected] 2Parks & Wildlife Division, Dept of Environment and Conservation (NSW), PO Box 375 Kurnell NSW 2231 AUSTRALIA email [email protected] Abstract: The first scientific observations on the flora of eastern Australia were made at Botany Bay in April–May 1770. We discuss the landscapes of Botany Bay and particularly of the historic landing place at Kurnell (lat 34˚ 00’ S, long 151˚ 13’ E) (about 16 km south of central Sydney), as described in the journals of Lieutenant James Cook and Joseph Banks on the Endeavour voyage in 1770. We list 132 plant species that were collected at Botany Bay by Banks and Daniel Solander, the first scientific collections of Australian flora. The list is based on a critical assessment of unpublished lists compiled by authors who had access to the collection of the British Museum (now Natural History Museum), together with species from material at National Herbarium of New South Wales that has not been previously available. The list includes Bidens pilosa which has been previously regarded as an introduced species. In 1770 the Europeans set foot on Aboriginal land of the Dharawal people. Since that time the landscape has been altered in response to a succession of different land-uses; farming and grazing, commemorative tree planting, parkland planting, and pleasure ground and tourist visitation. -

Mangrove Guidebook for Southeast Asia
RAP PUBLICATION 2006/07 MANGROVE GUIDEBOOK FOR SOUTHEAST ASIA The designations and the presentation of material in this publication do not imply the expression of any opinion whatsoever on the part of the Food and Agriculture Organization of the United Nations concerning the legal status of any country, territory, city or area or of its frontiers or boundaries. The opinions expressed in this publication are those of the authors alone and do not imply any opinion whatsoever on the part of FAO. Authored by: Wim Giesen, Stephan Wulffraat, Max Zieren and Liesbeth Scholten ISBN: 974-7946-85-8 FAO and Wetlands International, 2006 Printed by: Dharmasarn Co., Ltd. First print: July 2007 For copies write to: Forest Resources Officer FAO Regional Office for Asia and the Pacific Maliwan Mansion Phra Atit Road, Bangkok 10200 Thailand E-mail: [email protected] ii FOREWORDS Large extents of the coastlines of Southeast Asian countries were once covered by thick mangrove forests. In the past few decades, however, these mangrove forests have been largely degraded and destroyed during the process of development. The negative environmental and socio-economic impacts on mangrove ecosystems have led many government and non- government agencies, together with civil societies, to launch mangrove conservation and rehabilitation programmes, especially during the 1990s. In the course of such activities, programme staff have faced continual difficulties in identifying plant species growing in the field. Despite a wide availability of mangrove guidebooks in Southeast Asia, none of these sufficiently cover species that, though often associated with mangroves, are not confined to this habitat. -

米埔的植物mai Po Plants
米埔的植物 Mai Po Plants 學名 英文名稱 中文名稱 Scientific Name English Name Chinese Name Acacia auriculiformis Ear-leaved Acacia 耳果相思 / 耳葉相思 Acacia confusa Taiwan Acacia 台灣相思 Acacia mangium Big-leaved Acacia 大葉相思 Acanthus ilicifolius Spiny Bears Breech 老鼠簕 Achyranthes aspera Common Achyranthes 土牛膝 / 倒扣草 Acrostichum aureum Leather Fern 鹵蕨 Aegiceras corniculatum - 蠟燭果 / 桐花樹 Ageratum conyzoides Billygoat-weed 藿香薊 Aglaia odorata Mock Lime 米仔蘭 Albizia lebbeck Lebbeck Tree 大葉合歡 Allamanda cathartica Allamanda 軟枝黃蟬 Alnus japonica Formosan Alder 赤楊 Alocasia macrorrhizos Giant Alocasia, Alocasia 海芋 Alpinia zerumbet Variegated Shell Ginger 花葉豔山薑 Alternanthera paronychioides - 星星蝦鉗菜 Alternanthera philoxeroides Alligator-weed 空心莧 / 空心蓮子草 Alternanthera sessilis Sessile Alternanthera 蝦鉗菜 / 蓮子草 Alysicarpus vaginalis Alyce Clover 鏈莢豆 Amaranthus viridis Green Amaranth 綠莧 / 野莧 Annona squamosa Sugar-apple 番荔枝 Antirhea chinensis Chinese Antirhea 毛茶 Apluda mutica Glutene-rice Grass 水蔗草 Aporusa dioica Aporusa, Common Aporosa 銀柴 Ardisia crenata Hilo Holly 大羅傘 Ardisia elliptica - 東方紫金牛 Ardisia lindleyana Spotted Ardisia 山血丹 / 腺點紫金牛 Artocarpus heterophyllus Jackfruit 菠蘿蜜 Asclepias curassavica Blood-flower 馬利筋/連生桂子花 Asparagus cochinchinensis Wild Asparagus 天門冬 Aster subulatus - 鑽形紫菀 Asystasia micrantha - 小花十萬錯 Atalantia buxifolia Box-leaved Atalantia 酒餅簕 Aucuba chinensis Chinese Aucuba 桃葉珊瑚 Avicennia marina Black Mangrove 海欖雌 / 白骨壤 Axonopus compressus Carpet Grass 地毯草 Bacopa monnieri Water hyssop 假馬齒莧 Last update: February 2018 最後更新日期:2018 年 6 月 學名 英文名稱 中文名稱 Scientific Name -

MANGROVES in HONAVAR FOREST DIVISION, UTTARA KANNADA The
MANGROVES IN HONAVAR FOREST DIVISION, UTTARA KANNADA The Uttara Kannada District Sea Board lies between 74° 9’ to 75°10’ east longitude and 13° 55’ to 15° 31’ north latitude and extends over an area of 10,327 Sq. km. The district has three main and distinctive regions: the Coastlands, the Sahyadri range and Eastern margin where the tableland begins. The coastlines are the best-developed area with a high degree of economic development and a high density of population. It is in this region, Karwar, Ankola, Kumta, Honavar and Bhatkal taluks are located. The five important rivers of Uttara Kannada are Kalinadi, Gangavali, Aghanashini Sharavathi and Venktapur. The mangrove zone towards the river mouth, having higher salinity, is known as ‘euhaline’. Along the coast of Uttara Kannada, the river mouths are rocky and with strong wave action, and not suitable for mangroves. This is unlike the deltaic river mouths of the east coast, where mangroves can grow luxuriantly. The euhaline zone has salinity range from 30 ppt to 40 ppt. The zone behind is called ‘polyhaline’ where the wave action is less and salinity ranges from 18 ppt to 30 ppt. The substratum is sandy clay. This region is ideal for mangroves like Sonneratia alba, Rhizophora spp., Avicennia spp., Bruguiera spp., Acanthus ilicifolius etc. The third zone, still behind is ‘mesohaline’ where salinity ranges from 5 ppt to 18 ppt. It has silty clay bottom and feeble wave action. The mangroves of this zone are Kandelia candel, A. ilicifolius, Rhizophora spp., Aegiceras corniculatum, S. alba etc. (Rao and Suresh, 2001). -

Propagule Dispersal Determines Mangrove Zonation at Intertidal and Estuarine Scales
Article Propagule Dispersal Determines Mangrove Zonation at Intertidal and Estuarine Scales Wenqing Wang, Xiaofei Li and Mao Wang * Key Laboratory of the Coastal and Wetland Ecosystems, Ministry of Education, College of the Environment & Ecology, Xiamen University, Xiamen 361102, China; [email protected] (W.W.); [email protected] (X.L.) * Correspondence: [email protected]; Tel.: +86-136-6603-7893 Received: 1 February 2019; Accepted: 7 March 2019; Published: 10 March 2019 Abstract: Propagule dispersal has generally been recognized as a vital factor affecting the spatial structure of tropical forest plants. However, available research shows that this hypothesis does not apply to mangrove species the propagules of which are dispersed by water. Due to the lack of comprehensive and quantitative information as well as the high spatio-temporal heterogeneity of the mangrove environment, the exact factors affecting the spatial structure of mangrove forests are poorly understood. To assess this, we selected a mangrove estuary with high mangrove species richness that experiences great changes in water salinity. After investigating the zonation of mature mangrove individuals across tides and the estuary, we measured the size and initial specific gravity of the propagules and then selected the eight most common species from which to observe the changes in specific gravity, buoyancy, and root initiation during dispersal at different sites with different water salinity regimes. The relationships among distribution patterns, propagule establishment, and dispersal behavior were investigated. We found that mangrove propagule dispersal is not a passively buoyant process controlled by water currents. During dispersal, mangrove propagules can actively adjust their specific gravity and root initiation. -

2. AEGICERAS Gaertner, Fruct. & Sem. 1: 216. L788
Flora of China 15: 9. 1996. 2. AEGICERAS Gaertner, Fruct. & Sem. 1: 216. l788. 蜡烛果属 la zhu guo shu Shrubs or small trees. Leaves alternate or subopposite. Inflorescences terminal or rarely axillary, umbellate. Flowers bisexual, 5-merous. Corolla campanulate, united into a tube; lobes ovate or ovate-lanceolate, imbricate, overlapping to right in bud, recurved or reflexed at anthesis, not glandular. Basal part of filaments united into a tube as long as corolla tube, distal part free, exserted; anthers ovate, 2-celled, dehiscing longitudinally, transversely septate. Ovary superior; ovules numerous, within a globose placenta. Style elongated; stigma apiculate. Fruit elongated, terete, curved, 1-seeded capsules; exocarp dry, crustaceous, dehiscing by a longitudinal fissure or separating into 2 fragments along back and front; endocarp somewhat fleshy; persistent calyx compactly enclosing fruit base. Seeds occupying whole cavity; embryo terete, curved. Two species: India, Malaysia, Philippines, Sri Lanka, Vietnam, and Australia; one species in China. 1. Aegiceras corniculatum (Linnaeus) Blanco, Fl. Filip. 79. 1837. 蜡烛果 la zhu guo Rhizophora corniculata Linnaeus, Amoen. Acad. 4: l23. l759; Aegiceras majus Gaertner; Umbraculum corniculatum (Linnaeus) Kuntze. Plants ca. 1.5 m tall. Branchlets reddish or blackish brown, glabrous. Petiole 5–10 mm; leaf blade elliptic or broadly obo- vate, 3–8 × 2–4.5 cm, leathery, base cuneate, margin entire, revolute, apex rounded or emarginate; lateral veins 7–11 on each side of midrib, densely puberulent abaxially. Inflores- cences terminal, subsessile, ca. 10-flowered; bracts oblong- lanceolate, deciduous. Pedicel ca. 1 cm, ± glandular. Flowers ca. 1 cm. Calyx lobes rhomboid, 5–6 × 3–4 mm, leathery, prominently veined, black glandular at base, asymmetric, entire, apex broadly rounded. -
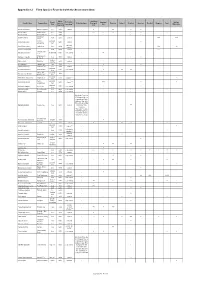
Appendix 8.2 Flora Species Recorded with the Assessment Area
Appendix 8.2 Flora Species Recorded with the Assessment Area Native / Project Area: Growth Distribution in Developed Modified Scientific Name Common Name Exotic to Protection Status Developed Plantation Orchard Shrubland Grassland Reedbed Mangrove Pond Form Hong Kong (1) Area Watercourse Hong Kong Area Acacia auriculiformis Ear-leaved Acacia tree exotic common - + ++ + + Acacia confusa Taiwan Acacia tree exotic - - +++ ++ +++ + ++ + Acacia mangium Big-leaved Acacia tree exotic - - + + + Spiny Bears Acanthus ilicifolius shrub native common - +++ +++ Breech Common perennial Achyranthes aspera native common - + Achyranthes herb restricted; Acrostichum aureum Leather Fern herb native - +++ ++ common (2) Aegiceras corniculatum - shrub native common - +++ Sensitive Joint- Aeschynomene indica shrubby herb native very common - ++ vetch Billygoat-weed , Ageratum conyzoides herb exotic common - + ++ Goatweed shrub or Aglaia odorata Mock Lime exotic common - + small tree Albizia lebbeck Lebbeck Tree tree exotic - - + + Aleurites moluccana Candlenut Tree tree exotic common - ++ perennial Alocasia macrorrhizos Giant Alocasia native very common - + + ++ ++ herb Chinese Aloe , perennial Aloe vera var. chinensis exotic - - + + Barbados Aloe herb perennial Alternanthera philoxeroides Alligator-weed exotic common (2) - + herb Sessile perennial Alternanthera sessilis native common (2) - +++ + + Alternanthera herb perennial Alysicarpus vaginalis Alyce Clover native very common - + + herb Amaranthus viridis Green Amaranth herb native very common - + + + Aporusa -

"True Mangroves" Plant Species Traits
Biodiversity Data Journal 5: e22089 doi: 10.3897/BDJ.5.e22089 Data Paper Dataset of "true mangroves" plant species traits Aline Ferreira Quadros‡‡, Martin Zimmer ‡ Leibniz Centre for Tropical Marine Research, Bremen, Germany Corresponding author: Aline Ferreira Quadros ([email protected]) Academic editor: Luis Cayuela Received: 06 Nov 2017 | Accepted: 29 Nov 2017 | Published: 29 Dec 2017 Citation: Quadros A, Zimmer M (2017) Dataset of "true mangroves" plant species traits. Biodiversity Data Journal 5: e22089. https://doi.org/10.3897/BDJ.5.e22089 Abstract Background Plant traits have been used extensively in ecology. They can be used as proxies for resource-acquisition strategies and facilitate the understanding of community structure and ecosystem functioning. However, many reviews and comparative analysis of plant traits do not include mangroves plants, possibly due to the lack of quantitative information available in a centralised form. New information Here a dataset is presented with 2364 records of traits of "true mangroves" species, gathered from 88 references (published articles, books, theses and dissertations). The dataset contains information on 107 quantitative traits and 18 qualitative traits for 55 species of "true mangroves" (sensu Tomlinson 2016). Most traits refer to components of living trees (mainly leaves), but litter traits were also included. Keywords Mangroves, Rhizophoraceae, leaf traits, plant traits, halophytes © Quadros A, Zimmer M. This is an open access article distributed under the terms of the Creative Commons Attribution License (CC BY 4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. 2 Quadros A, Zimmer M Introduction The vegetation of mangrove forests is loosely classified as "true mangroves" or "mangrove associates". -

The Role of Freezing in Setting the Latitudinal Limits of Mangrove Forests
UC Berkeley UC Berkeley Previously Published Works Title The role of freezing in setting the latitudinal limits of mangrove forests Permalink https://escholarship.org/uc/item/7wp3h4gx Journal New Phytologist, 173(3) ISSN 0028-646X Authors Stuart, S A Choat, B Martin, K C et al. Publication Date 2007 Peer reviewed eScholarship.org Powered by the California Digital Library University of California Stuart et al. The latitudinal limits of mangrove forests 1 Title: The role of freezing in setting the latitudinal limits of mangrove forests 2 S. A. Stuart1,2, B. Choat1,3, K. Martin,4 N. M. Holbrook1 and M. C. Ball4 3 1 Department of Organismic and Evolutionary Biology, Harvard University, Biological 4 Laboratories, 16 Divinity Avenue, Cambridge, Massachusetts, 02138, USA 5 2 Current address: Department of Integrative Biology, 3960 Valley Life Sciences Building 6 #3140, University of California, Berkeley, CA 94702, USA 7 3 Current address: Department of Viticulture and Enology, University of California, Davis, 8 California, 95616, USA 9 4 Ecosystem Dynamics Group, Research School of Biological Sciences, Institute of Advanced 10 Studies, Australian National University, Canberra, A.C.T., 0200, Australia. 11 12 Corresponding Author: Stephanie Stuart 13 14 Department of Integrative Biology 15 3060 VLSB #3140 16 University of California, Berkeley 17 Berkeley, CA 94720-3140 18 USA 19 Phone: +1 510 643 5340 20 Fax: +1 510 643 6264 21 Email: [email protected] 22 23 Word counts 24 Total word count: 4,746 25 Summary word count: 171 26 Introduction word count: 661 27 Methods word count: 1,250 28 Results word count: 526 29 Discussion word count: 925 30 Tables: 2 31 Figures: 2 32 33 Abbreviations: 34 EDX, Energy Dispersive X-ray Microanalysis 35 PLC, Percent Loss of Conductivity 36 Page 1 of 22 Stuart et al.