(12) Patent Application Publication (10) Pub. No.: US 2010/0159007 A1 Staniforth (43) Pub
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-
![Doxapram Shortens Recovery Following Sevoflurane Anesthesia [Le Doxapram Hâte La Récupération Après Une Anesthésie Au Sévoflurane]](https://docslib.b-cdn.net/cover/1653/doxapram-shortens-recovery-following-sevoflurane-anesthesia-le-doxapram-h%C3%A2te-la-r%C3%A9cup%C3%A9ration-apr%C3%A8s-une-anesth%C3%A9sie-au-s%C3%A9voflurane-391653.webp)
Doxapram Shortens Recovery Following Sevoflurane Anesthesia [Le Doxapram Hâte La Récupération Après Une Anesthésie Au Sévoflurane]
456 CANADIAN JOURNALGENERAL OF ANESTHESIA ANESTHESIA Doxapram shortens recovery following sevoflurane anesthesia [Le doxapram hâte la récupération après une anesthésie au sévoflurane] Chi-Chen Wu MD,* Martin S. Mok MD,* Jui-Yuan Chen MD,† Gong-Jhe Wu MD,† Yeong-Ray Wen MD,† Chao-Shun Lin MD* Purpose: A randomized, double blind controlled trial was possibilité de serrer la main sur demande, le temps d’extubation et undertaken to investigate the effect of doxapram on recovery le score de récupération d’Aldrete. Les valeurs de l’index bispectral, times and bispectral index following sevoflurane anesthesia. la tension artérielle systolique et la fréquence cardiaque ont été Methods: Upon completion of surgery under sevoflurane anes- enregistrées avant l’anesthésie, pendant l’opération et à chaque thesia, 60 adult patients were randomly allocated to receive minute pendant 15 min après l’administration du médicament. either doxapram hydrochloride 1 mg·kg–1 iv or saline placebo. Résultats : Le temps écoulé avant l’ouverture des yeux a été plus Clinical recovery from anesthesia was assessed by time to eye court avec le doxapram qu’avec le placebo (6,9 ± 2,2 min vs 9,9 opening on verbal command, hand squeezing on command, ± 3,1 min, P < 0,05). Les scores moyens de l’index bispectral ont time to extubation, and the Aldrete recovery score. Bispectral été aussi plus élevés avec le doxapram sept à huit minutes après index values, systolic blood pressure, and heart rate were l’administration du médicament expérimental (P < 0,05). Un recorded at baseline (before anesthesia), during surgery, and retour à la conscience plus rapide a été associé à une plus grande every minute for 15 min after administration of the study drug. -

“Inactive” Ingredients in Pharmaceutical Products: Update (Subject Review)
AMERICAN ACADEMY OF PEDIATRICS Committee on Drugs “Inactive” Ingredients in Pharmaceutical Products: Update (Subject Review) ABSTRACT. Because of an increasing number of re- bronchospasm from antiasthmatic drugs, aspartame- ports of adverse reactions associated with pharmaceutical induced headache and seizures, saccharin-induced excipients, in 1985 the Committee on Drugs issued a cross-sensitivity reactions in children with sulfon- position statement1 recommending that the Food and amide allergy, benzyl alcohol toxicity in neonates Drug Administration mandate labeling of over-the- receiving high-dose continuous infusion with pre- counter and prescription formulations to include a qual- served medications, dye-related cross-reactions in itative list of inactive ingredients. However, labeling of inactive ingredients remains voluntary. Adverse reac- children with aspirin intolerance, lactose-induced di- tions continue to be reported, although some are no arrhea, and propylene glycol-induced hyperosmola- longer considered clinically significant, and other new lity and lactic acidosis. Although many other excipi- reactions have emerged. The original statement, there- ents have been implicated in causing adverse fore, has been updated and its information expanded. reactions, these are the most significant in the pedi- atric population. ABBREVIATIONS. FDA, Food and Drug Administration; MDIs, metered-dose inhalers ANTIASTHMATIC MEDICATIONS It is readily appreciated that some percentage of asthmatic children will develop a “paradoxical” Pharmaceutical products often contain agents that bronchospasm after they inhale their medication. Be- have a variety of purposes, including improvement cause many of these reactions were attributed to of the appearance, bioavailability, stability, and pal- sulfite, which had been highly publicized as a caus- atability of the product. Excipients (substances ative agent, it was often first suspected. -

Doxapram Elisa Kit Instructions Product #106219 & 106216 Forensic Use Only
Neogen Corporation 944 Nandino Blvd., Lexington KY 40511 USA 800/477-8201 USA/Canada | 859/254-1221 Fax: 859/255-5532 | E-mail: [email protected] | Web: www.neogen.com/Toxicology DOXAPRAM ELISA KIT INSTRUCTIONS PRODUCT #106219 & 106216 FORENSIC USE ONLY INTENDED USE: For the determination of trace quantities of Doxapram and/or other metabolites in human urine, blood, oral fluid. DESCRIPTION Neogen Corporation’s Doxapram ELISA (Enzyme-Linked ImmunoSorbent Assay) test kit is a qualitative one-step kit designed for use as a screening device for the detection of drugs and/or their metabolites. The kit was designed for screening purposes and is intended for forensic use only. It is recommended that all suspect samples be confirmed by a quantitative method such as gas chromatography/mass spectrometry (GC/MS). ASSAY PRINCIPLES Neogen Corporation’s test kit operates on the basis of competition between the drug or its metabolite in the sample and the drug-enzyme conjugate for a limited number of antibody binding sites. First, the sample or control is added to the microplate. Next, the diluted drug-enzyme conjugate is added and the mixture is incubated at room temperature. During this incubation, the drug in the sample or the drug-enzyme conjugate binds to antibody immobilized in the microplate wells. After incubation, the plate is washed 3 times to remove any unbound sample or drug-enzyme conjugate. The presence of bound drug-enzyme conjugate is recognized by the addition of K-Blue® Substrate (TMB). After a 30 minute substrate incubation, the reaction is halted with the addition of Red Stop Solution. -

Neonatal Medications & Nutrition
Neonatal Medications & Nutrition A Comprehensive Guide 3rd Edition KARIN E. ZENK, PHARMD, FASHP Associate Clinical Professor of Pediatrics, University of California, Irvine, and in private consulting practice Formerly, Pharmacist-Specialist in Pediatrics and Neonatology, University of California, Irvine Medical Center JACK H. SILLS, MD Medical Director, Neonatal Intensive Care Unit, University of California, Irvine Medical Center, Clinical Professor of Pediatrics, University of California, Irvine ROBIN M. KOEPPEL, RNC, MS, CPNP, CS Neonatal Clinical Nurse Specialist/Pediatric Nurse Practitioner, University of California, Irvine Medical Center NK BOOK PUBLISHERS SAN T-A ROSA, CALIFORNIA Neonatal Medications & Nutrition TABLE OF CONTENTS Table of Contents by Therapeutic Categories and Indications xv Notice xxvi How to Use This Book xxvii Special Considerations in Neonatal Drug Therapy xxix SECTION I-DRUC MONOGRAPHS Acetaminophen (Tylenol) 1 Acetylcysteine (Mucomyst) 8 Acyclovir (Zovirax) 11 Adenosine (Adenocard) 13 Albumin 15 Albuterol (Proventil, Ventolin) .17 Alprostadil (Prostin VR Pediatric) 22 Alteplase, Recombinant (Activase, Cathflo Activase, Actilyse) 25 Aluminum and Magnesium Hydroxides (Maalox) 33 Amikacin (Amikin) 35 Aminocaproic Acid (Amicar) 37 Aminophylline 40 Amiodarone (Cordarone) 45 Amoxicillin (Amoxil) 53 Amphotericin B, Conventional (Fungizone) and Amphotericin B Lipid Based (Abelcet, Amphotec, AmBisome) 55 Ampicillin 60 Aquaphor Ointment 63 Arginine Hydrochloride (R-Gene 10) 66 Atraeurium (Tracrium) 75 Atropine 78 Aztreonam -
![Ehealth DSI [Ehdsi V2.2.2-OR] Ehealth DSI – Master Value Set](https://docslib.b-cdn.net/cover/8870/ehealth-dsi-ehdsi-v2-2-2-or-ehealth-dsi-master-value-set-1028870.webp)
Ehealth DSI [Ehdsi V2.2.2-OR] Ehealth DSI – Master Value Set
MTC eHealth DSI [eHDSI v2.2.2-OR] eHealth DSI – Master Value Set Catalogue Responsible : eHDSI Solution Provider PublishDate : Wed Nov 08 16:16:10 CET 2017 © eHealth DSI eHDSI Solution Provider v2.2.2-OR Wed Nov 08 16:16:10 CET 2017 Page 1 of 490 MTC Table of Contents epSOSActiveIngredient 4 epSOSAdministrativeGender 148 epSOSAdverseEventType 149 epSOSAllergenNoDrugs 150 epSOSBloodGroup 155 epSOSBloodPressure 156 epSOSCodeNoMedication 157 epSOSCodeProb 158 epSOSConfidentiality 159 epSOSCountry 160 epSOSDisplayLabel 167 epSOSDocumentCode 170 epSOSDoseForm 171 epSOSHealthcareProfessionalRoles 184 epSOSIllnessesandDisorders 186 epSOSLanguage 448 epSOSMedicalDevices 458 epSOSNullFavor 461 epSOSPackage 462 © eHealth DSI eHDSI Solution Provider v2.2.2-OR Wed Nov 08 16:16:10 CET 2017 Page 2 of 490 MTC epSOSPersonalRelationship 464 epSOSPregnancyInformation 466 epSOSProcedures 467 epSOSReactionAllergy 470 epSOSResolutionOutcome 472 epSOSRoleClass 473 epSOSRouteofAdministration 474 epSOSSections 477 epSOSSeverity 478 epSOSSocialHistory 479 epSOSStatusCode 480 epSOSSubstitutionCode 481 epSOSTelecomAddress 482 epSOSTimingEvent 483 epSOSUnits 484 epSOSUnknownInformation 487 epSOSVaccine 488 © eHealth DSI eHDSI Solution Provider v2.2.2-OR Wed Nov 08 16:16:10 CET 2017 Page 3 of 490 MTC epSOSActiveIngredient epSOSActiveIngredient Value Set ID 1.3.6.1.4.1.12559.11.10.1.3.1.42.24 TRANSLATIONS Code System ID Code System Version Concept Code Description (FSN) 2.16.840.1.113883.6.73 2017-01 A ALIMENTARY TRACT AND METABOLISM 2.16.840.1.113883.6.73 2017-01 -

Jp Xvii the Japanese Pharmacopoeia
JP XVII THE JAPANESE PHARMACOPOEIA SEVENTEENTH EDITION Official from April 1, 2016 English Version THE MINISTRY OF HEALTH, LABOUR AND WELFARE Notice: This English Version of the Japanese Pharmacopoeia is published for the convenience of users unfamiliar with the Japanese language. When and if any discrepancy arises between the Japanese original and its English translation, the former is authentic. The Ministry of Health, Labour and Welfare Ministerial Notification No. 64 Pursuant to Paragraph 1, Article 41 of the Law on Securing Quality, Efficacy and Safety of Products including Pharmaceuticals and Medical Devices (Law No. 145, 1960), the Japanese Pharmacopoeia (Ministerial Notification No. 65, 2011), which has been established as follows*, shall be applied on April 1, 2016. However, in the case of drugs which are listed in the Pharmacopoeia (hereinafter referred to as ``previ- ous Pharmacopoeia'') [limited to those listed in the Japanese Pharmacopoeia whose standards are changed in accordance with this notification (hereinafter referred to as ``new Pharmacopoeia'')] and have been approved as of April 1, 2016 as prescribed under Paragraph 1, Article 14 of the same law [including drugs the Minister of Health, Labour and Welfare specifies (the Ministry of Health and Welfare Ministerial Notification No. 104, 1994) as of March 31, 2016 as those exempted from marketing approval pursuant to Paragraph 1, Article 14 of the Same Law (hereinafter referred to as ``drugs exempted from approval'')], the Name and Standards established in the previous Pharmacopoeia (limited to part of the Name and Standards for the drugs concerned) may be accepted to conform to the Name and Standards established in the new Pharmacopoeia before and on September 30, 2017. -
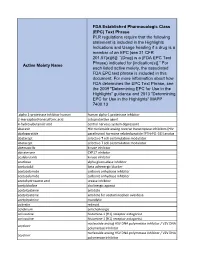
Active Moiety Name FDA Established Pharmacologic Class (EPC) Text
FDA Established Pharmacologic Class (EPC) Text Phrase PLR regulations require that the following statement is included in the Highlights Indications and Usage heading if a drug is a member of an EPC [see 21 CFR 201.57(a)(6)]: “(Drug) is a (FDA EPC Text Phrase) indicated for [indication(s)].” For Active Moiety Name each listed active moiety, the associated FDA EPC text phrase is included in this document. For more information about how FDA determines the EPC Text Phrase, see the 2009 "Determining EPC for Use in the Highlights" guidance and 2013 "Determining EPC for Use in the Highlights" MAPP 7400.13. .alpha. -

Current Awareness in Clinical Toxicology Editors: Damian Ballam Msc and Allister Vale MD
Current Awareness in Clinical Toxicology Editors: Damian Ballam MSc and Allister Vale MD May 2016 CONTENTS General Toxicology 8 Metals 33 Management 16 Pesticides 35 Drugs 19 Chemical Warfare 37 Chemical Incidents & 28 Plants 37 Pollution Chemicals 29 Animals 38 CURRENT AWARENESS PAPERS OF THE MONTH Efficacy and effectiveness of anti-digoxin antibodies in chronic digoxin poisonings from the DORA study (ATOM-1) Chan BS, Isbister GK, O'Leary M, Chiew A, Buckley NA. Clin Toxicol 2016; online early: doi: 10.1080/15563650.2016.1175620: Context We hypothesized that in chronic digoxin toxicity, anti-digoxin antibodies (Fab) would be efficacious in binding digoxin, but this may not translate into improved clinical outcomes. Objective This study aims to investigate changes in free digoxin concentrations and clinical effects on heart rate and potassium concentrations in chronic digoxin poisoning when anti-digoxin Fab are given. Materials and methods This is a prospective observational study. Patients were recruited if they have been treated with anti-digoxin Fab for chronic digoxin poisoning. Data was entered into a standardised prospective form, supplemented with medical records. Their serum or plasma was collected, analysed for free and bound digoxin and free anti-digoxin Fab concentrations. Results From September 2013 to February 2015, 36 patients (median age, 78 years; 22 females) Current Awareness in Clinical Toxicology is produced monthly for the American Academy of Clinical Toxicology by the Birmingham Unit of the UK National Poisons Information Service, with contributions from the Cardiff, Edinburgh, and Newcastle Units. The NPIS is commissioned by Public Health England 2 were recruited from 18 hospitals. -

Anatomical Classification Guidelines V2021 EPHMRA ANATOMICAL CLASSIFICATION GUIDELINES 2021
EPHMRA ANATOMICAL CLASSIFICATION GUIDELINES 2021 Anatomical Classification Guidelines V2021 "The Anatomical Classification of Pharmaceutical Products has been developed and maintained by the European Pharmaceutical Marketing Research Association (EphMRA) and is therefore the intellectual property of this Association. EphMRA's Classification Committee prepares the guidelines for this classification system and takes care for new entries, changes and improvements in consultation with the product's manufacturer. The contents of the Anatomical Classification of Pharmaceutical Products remain the copyright to EphMRA. Permission for use need not be sought and no fee is required. We would appreciate, however, the acknowledgement of EphMRA Copyright in publications etc. Users of this classification system should keep in mind that Pharmaceutical markets can be segmented according to numerous criteria." © EphMRA 2021 Anatomical Classification Guidelines V2021 CONTENTS PAGE INTRODUCTION A ALIMENTARY TRACT AND METABOLISM 1 B BLOOD AND BLOOD FORMING ORGANS 28 C CARDIOVASCULAR SYSTEM 36 D DERMATOLOGICALS 51 G GENITO-URINARY SYSTEM AND SEX HORMONES 58 H SYSTEMIC HORMONAL PREPARATIONS (EXCLUDING SEX HORMONES) 68 J GENERAL ANTI-INFECTIVES SYSTEMIC 72 K HOSPITAL SOLUTIONS 88 L ANTINEOPLASTIC AND IMMUNOMODULATING AGENTS 96 M MUSCULO-SKELETAL SYSTEM 106 N NERVOUS SYSTEM 111 P PARASITOLOGY 122 R RESPIRATORY SYSTEM 124 S SENSORY ORGANS 136 T DIAGNOSTIC AGENTS 143 V VARIOUS 145 Anatomical Classification Guidelines V2021 INTRODUCTION The Anatomical Classification was initiated in 1971 by EphMRA. It has been developed jointly by Intellus/PBIRG and EphMRA. It is a subjective method of grouping certain pharmaceutical products and does not represent any particular market, as would be the case with any other classification system. -

Pharmacy Data Management Drug Exception List
Pharmacy Data Management Drug Exception List Patch PSS*1*127 updated the following drugs with the listed NCPDP Multiplier and NCPDP Dispense Unit. These two fields were added as part of this patch to the DRUG file (#50). Please refer to the Release notes for ePharmacy/ECME Enhancements for Pharmacy Release Notes (BPS_1_5_EPHARMACY_RN_0907.PDF) on the VistA Documentation Library (VDL). The IEN column reflects the IEN for the VA PRODUCT file (#50.68). The ePharmacy Change Control Board provided the following list of drugs with the specified NCPDP Multiplier and NCPDP Dispense Unit values. This listing was used to update the DRUG file (#50) with a post install routine in the PSS*1*127 patch. NCPDP File 50.68 NCPDP Dispense IEN Product Name Multiplier Unit 2 ATROPINE SO4 0.4MG/ML INJ 1.00 ML 3 ATROPINE SO4 1% OINT,OPH 3.50 GM 6 ATROPINE SO4 1% SOLN,OPH 1.00 ML 7 ATROPINE SO4 0.5% OINT,OPH 3.50 GM 8 ATROPINE SO4 0.5% SOLN,OPH 1.00 ML 9 ATROPINE SO4 3% SOLN,OPH 1.00 ML 10 ATROPINE SO4 2% SOLN,OPH 1.00 ML 11 ATROPINE SO4 0.1MG/ML INJ 1.00 ML 12 ATROPINE SO4 0.05MG/ML INJ 1.00 ML 13 ATROPINE SO4 0.4MG/0.5ML INJ 1.00 ML 14 ATROPINE SO4 0.5MG/ML INJ 1.00 ML 15 ATROPINE SO4 1MG/ML INJ 1.00 ML 16 ATROPINE SO4 2MG/ML INJ 1.00 ML 18 ATROPINE SO4 2MG/0.7ML INJ 0.70 ML 21 ATROPINE SO4 0.3MG/ML INJ 1.00 ML 22 ATROPINE SO4 0.8MG/ML INJ 1.00 ML 23 ATROPINE SO4 0.1MG/ML INJ,SYRINGE,5ML 5.00 ML 24 ATROPINE SO4 0.1MG/ML INJ,SYRINGE,10ML 10.00 ML 25 ATROPINE SO4 1MG/ML INJ,AMP,1ML 1.00 ML 26 ATROPINE SO4 0.2MG/0.5ML INJ,AMP,0.5ML 0.50 ML 30 CODEINE PO4 30MG/ML -

Respiratory Stimulant Drugs in the Post-Operative Setting
Respiratory Physiology & Neurobiology 189 (2013) 395–402 View metadata, citation and similar papers at core.ac.uk brought to you by CORE Contents lists available at ScienceDirect provided by Elsevier - Publisher Connector Respiratory Physiology & Neurobiology j ournal homepage: www.elsevier.com/locate/resphysiol Review ଝଝ Respiratory stimulant drugs in the post-operative setting ∗ Francis J. Golder , Matthew M. Hewitt, James F. McLeod Galleon Pharmaceuticals, Inc., 213 Witmer Road, Horsham, PA 19044, USA a r t i c l e i n f o a b s t r a c t Article history: Drug-induced respiratory depression (DIRD) is a common problem encountered post-operatively and Accepted 11 June 2013 can persist for days after surgery. It is not always possible to predict the timing or severity of DIRD due to the number of contributing factors. A safe and effective respiratory stimulant could improve Keywords: patient care by avoiding the use of reversal agents (e.g., naloxone, which reverses analgesia as well as Surgery respiratory depression) thereby permitting better pain management by enabling the use of higher doses Ventilation of analgesics, facilitate weaning from prolonged ventilation, and ameliorate sleep-disordered breathing Anesthesia peri-operatively. The purpose of this review is to discuss the current pharmaceutical armamentarium Analeptic Opioid of drugs (doxapram and almitrine) that are licensed for use in humans as respiratory stimulants and Hypoxia that could be used to reverse drug-induced respiratory depression in the post-operative period. We also discuss new chemical entities (AMPAkines and GAL-021) that have been recently evaluated in Phase 1 clinical trials and where the initial regulatory registration would be as a respiratory stimulant. -

FDA Listing of Established Pharmacologic Class Text Phrases January 2021
FDA Listing of Established Pharmacologic Class Text Phrases January 2021 FDA EPC Text Phrase PLR regulations require that the following statement is included in the Highlights Indications and Usage heading if a drug is a member of an EPC [see 21 CFR 201.57(a)(6)]: “(Drug) is a (FDA EPC Text Phrase) indicated for Active Moiety Name [indication(s)].” For each listed active moiety, the associated FDA EPC text phrase is included in this document. For more information about how FDA determines the EPC Text Phrase, see the 2009 "Determining EPC for Use in the Highlights" guidance and 2013 "Determining EPC for Use in the Highlights" MAPP 7400.13.