Standard Sequencing Service Data File Formats File Format V2.4 Software V2.4 December 2012
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Supplemental Note Hominoid Fission of Chromosome 14/15 and Role Of
Supplemental Note Supplemental Note Hominoid fission of chromosome 14/15 and role of segmental duplications Giuliana Giannuzzi, Michele Pazienza, John Huddleston, Francesca Antonacci, Maika Malig, Laura Vives, Evan E. Eichler and Mario Ventura 1. Analysis of the macaque contig spanning the hominoid 14/15 fission site We grouped contig clones based on their FISH pattern on human and macaque chromosomes (Groups F1, F2, F3, G1, G2, G3, and G4). Group F2 clones (Hsa15b- and Hsa15c-positive) showed a single signal on macaque 7q and three signal clusters on human chromosome 15: at the orthologous 15q26, as expected, as well as at 15q11–14 and 15q24–25, which correspond to the actual and ancestral pericentromeric regions, respectively (Ventura et al. 2003). Indeed, this locus contains an LCR15 copy (Pujana et al. 2001) in both the macaque and human genomes. Most BAC clones were one-end anchored in the human genome (chr15:100,028–100,071 kb). In macaque this region experienced a 64 kb duplicative insertion from chromosome 17 (orthologous to human chromosome 13) (Figure 2), with the human configuration (absence of insertion) likely being the ancestral state because it is identical in orangutan and marmoset. Four Hsa15b- positive clones mapped on macaque and human chromosome 19, but neither human nor macaque assemblies report the STS Hsa15b duplicated at this locus. The presence of assembly gaps in macaque may explain why the STS is not annotated in this region. We aligned the sequence of macaque CH250-70H12 (AC187495.2) versus its human orthologous sequence (hg18 chr15:100,039k-100,170k) and found a 12 kb human expansion through tandem duplication of a ~100 bp unit corresponding to a portion of exon 20 of DNM1 (Figure S3). -

Genetic Variation Across the Human Olfactory Receptor Repertoire Alters Odor Perception
bioRxiv preprint doi: https://doi.org/10.1101/212431; this version posted November 1, 2017. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. Genetic variation across the human olfactory receptor repertoire alters odor perception Casey Trimmer1,*, Andreas Keller2, Nicolle R. Murphy1, Lindsey L. Snyder1, Jason R. Willer3, Maira Nagai4,5, Nicholas Katsanis3, Leslie B. Vosshall2,6,7, Hiroaki Matsunami4,8, and Joel D. Mainland1,9 1Monell Chemical Senses Center, Philadelphia, Pennsylvania, USA 2Laboratory of Neurogenetics and Behavior, The Rockefeller University, New York, New York, USA 3Center for Human Disease Modeling, Duke University Medical Center, Durham, North Carolina, USA 4Department of Molecular Genetics and Microbiology, Duke University Medical Center, Durham, North Carolina, USA 5Department of Biochemistry, University of Sao Paulo, Sao Paulo, Brazil 6Howard Hughes Medical Institute, New York, New York, USA 7Kavli Neural Systems Institute, New York, New York, USA 8Department of Neurobiology and Duke Institute for Brain Sciences, Duke University Medical Center, Durham, North Carolina, USA 9Department of Neuroscience, University of Pennsylvania School of Medicine, Philadelphia, Pennsylvania, USA *[email protected] ABSTRACT The human olfactory receptor repertoire is characterized by an abundance of genetic variation that affects receptor response, but the perceptual effects of this variation are unclear. To address this issue, we sequenced the OR repertoire in 332 individuals and examined the relationship between genetic variation and 276 olfactory phenotypes, including the perceived intensity and pleasantness of 68 odorants at two concentrations, detection thresholds of three odorants, and general olfactory acuity. -

Assembly and Annotation of an Ashkenazi Human Reference Genome
bioRxiv preprint doi: https://doi.org/10.1101/2020.03.18.997395; this version posted March 18, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. Assembly and Annotation of an Ashkenazi Human Reference Genome Alaina Shumate1,2,† Aleksey V. Zimin1,2,† Rachel M. Sherman1,3 Daniela Puiu1,3 Justin M. Wagner4 Nathan D. Olson4 Mihaela Pertea1,2 Marc L. Salit5 Justin M. Zook4 Steven L. Salzberg1,2,3,6* 1Center for Computational Biology, Johns Hopkins University, Baltimore, MD 2Department of Biomedical Engineering, Johns Hopkins University, Baltimore, MD 3Department of Computer Science, Johns Hopkins University, Baltimore, MD 4National Institute of Standards and Technology, Gaithersburg, MD 5Joint Initiative for Metrology in Biology, Stanford University, Stanford, CA 6Department of Biostatistics, Johns Hopkins University, Baltimore, MD †These authors contributed equally to this work. *Corresponding author. Email: [email protected] Abstract Here we describe the assembly and annotation of the genome of an Ashkenazi individual and the creation of a new, population-specific human reference genome. This genome is more contiguous and more complete than GRCh38, the latest version of the human reference genome, and is annotated with highly similar gene content. The Ashkenazi reference genome, Ash1, contains 2,973,118,650 nucleotides as compared to 2,937,639,212 in GRCh38. Annotation identified 20,157 protein-coding genes, of which 19,563 are >99% identical to their counterparts on GRCh38. Most of the remaining genes have small differences. -

Seq2pathway Vignette
seq2pathway Vignette Bin Wang, Xinan Holly Yang, Arjun Kinstlick May 19, 2021 Contents 1 Abstract 1 2 Package Installation 2 3 runseq2pathway 2 4 Two main functions 3 4.1 seq2gene . .3 4.1.1 seq2gene flowchart . .3 4.1.2 runseq2gene inputs/parameters . .5 4.1.3 runseq2gene outputs . .8 4.2 gene2pathway . 10 4.2.1 gene2pathway flowchart . 11 4.2.2 gene2pathway test inputs/parameters . 11 4.2.3 gene2pathway test outputs . 12 5 Examples 13 5.1 ChIP-seq data analysis . 13 5.1.1 Map ChIP-seq enriched peaks to genes using runseq2gene .................... 13 5.1.2 Discover enriched GO terms using gene2pathway_test with gene scores . 15 5.1.3 Discover enriched GO terms using Fisher's Exact test without gene scores . 17 5.1.4 Add description for genes . 20 5.2 RNA-seq data analysis . 20 6 R environment session 23 1 Abstract Seq2pathway is a novel computational tool to analyze functional gene-sets (including signaling pathways) using variable next-generation sequencing data[1]. Integral to this tool are the \seq2gene" and \gene2pathway" components in series that infer a quantitative pathway-level profile for each sample. The seq2gene function assigns phenotype-associated significance of genomic regions to gene-level scores, where the significance could be p-values of SNPs or point mutations, protein-binding affinity, or transcriptional expression level. The seq2gene function has the feasibility to assign non-exon regions to a range of neighboring genes besides the nearest one, thus facilitating the study of functional non-coding elements[2]. Then the gene2pathway summarizes gene-level measurements to pathway-level scores, comparing the quantity of significance for gene members within a pathway with those outside a pathway. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Targeted Exome Sequencing Provided Comprehensive Genetic Diagnosis of Congenital Anomalies of the Kidney and Urinary Tract
Journal of Clinical Medicine Article Targeted Exome Sequencing Provided Comprehensive Genetic Diagnosis of Congenital Anomalies of the Kidney and Urinary Tract 1,2, 3,4, 3 1,5 Yo Han Ahn y, Chung Lee y, Nayoung K. D. Kim , Eujin Park , Hee Gyung Kang 1,2,6,* , Il-Soo Ha 1,2,6, Woong-Yang Park 3,4,7 and Hae Il Cheong 1,2,6 1 Department of Pediatrics, Seoul National University College of Medicine, Seoul 03080, Korea; [email protected] (Y.H.A.); [email protected] (E.P.); [email protected] (I.-S.H.); [email protected] (H.I.C.) 2 Department of Pediatrics, Seoul National University Children’s Hospital, Seoul 03080, Korea 3 Samsung Genome Institute, Samsung Medical Center, Seoul 06351, Korea; [email protected] (C.L.); [email protected] (N.K.D.K.); [email protected] (W.-Y.P.) 4 Department of Health Sciences and Technology, Samsung Advanced Institute for Health Sciences and Technology, Sungkyunkwan University, Seoul 06351, Korea 5 Department of Pediatrics, Kangnam Sacred Heart Hospital, Hallym University College of Medicine, Seoul 07441, Korea 6 Kidney Research Institute, Medical Research Center, Seoul National University College of Medicine, Seoul 03080, Korea 7 Department of Molecular Cell Biology, Sungkyunkwan University School of Medicine, Suwon 16419, Korea * Correspondence: [email protected] These authors equally contributed to this article. y Received: 31 January 2020; Accepted: 8 March 2020; Published: 10 March 2020 Abstract: Congenital anomalies of the kidney and urinary tract (CAKUT) are the most common cause of chronic kidney disease in children. -

Noelia Díaz Blanco
Effects of environmental factors on the gonadal transcriptome of European sea bass (Dicentrarchus labrax), juvenile growth and sex ratios Noelia Díaz Blanco Ph.D. thesis 2014 Submitted in partial fulfillment of the requirements for the Ph.D. degree from the Universitat Pompeu Fabra (UPF). This work has been carried out at the Group of Biology of Reproduction (GBR), at the Department of Renewable Marine Resources of the Institute of Marine Sciences (ICM-CSIC). Thesis supervisor: Dr. Francesc Piferrer Professor d’Investigació Institut de Ciències del Mar (ICM-CSIC) i ii A mis padres A Xavi iii iv Acknowledgements This thesis has been made possible by the support of many people who in one way or another, many times unknowingly, gave me the strength to overcome this "long and winding road". First of all, I would like to thank my supervisor, Dr. Francesc Piferrer, for his patience, guidance and wise advice throughout all this Ph.D. experience. But above all, for the trust he placed on me almost seven years ago when he offered me the opportunity to be part of his team. Thanks also for teaching me how to question always everything, for sharing with me your enthusiasm for science and for giving me the opportunity of learning from you by participating in many projects, collaborations and scientific meetings. I am also thankful to my colleagues (former and present Group of Biology of Reproduction members) for your support and encouragement throughout this journey. To the “exGBRs”, thanks for helping me with my first steps into this world. Working as an undergrad with you Dr. -

Subterranean Mammals Show Convergent Regression in Ocular Genes and Enhancers, Along with Adaptation to Tunneling
RESEARCH ARTICLE Subterranean mammals show convergent regression in ocular genes and enhancers, along with adaptation to tunneling Raghavendran Partha1, Bharesh K Chauhan2,3, Zelia Ferreira1, Joseph D Robinson4, Kira Lathrop2,3, Ken K Nischal2,3, Maria Chikina1*, Nathan L Clark1* 1Department of Computational and Systems Biology, University of Pittsburgh, Pittsburgh, United States; 2UPMC Eye Center, Children’s Hospital of Pittsburgh, Pittsburgh, United States; 3Department of Ophthalmology, University of Pittsburgh School of Medicine, Pittsburgh, United States; 4Department of Molecular and Cell Biology, University of California, Berkeley, United States Abstract The underground environment imposes unique demands on life that have led subterranean species to evolve specialized traits, many of which evolved convergently. We studied convergence in evolutionary rate in subterranean mammals in order to associate phenotypic evolution with specific genetic regions. We identified a strong excess of vision- and skin-related genes that changed at accelerated rates in the subterranean environment due to relaxed constraint and adaptive evolution. We also demonstrate that ocular-specific transcriptional enhancers were convergently accelerated, whereas enhancers active outside the eye were not. Furthermore, several uncharacterized genes and regulatory sequences demonstrated convergence and thus constitute novel candidate sequences for congenital ocular disorders. The strong evidence of convergence in these species indicates that evolution in this environment is recurrent and predictable and can be used to gain insights into phenotype–genotype relationships. DOI: https://doi.org/10.7554/eLife.25884.001 *For correspondence: [email protected] (MC); [email protected] (NLC) Competing interests: The Introduction authors declare that no The subterranean habitat has been colonized by numerous animal species for its shelter and unique competing interests exist. -
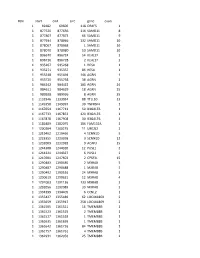
Chr Start End Size Gene Exon 1 69482 69600 118 OR4F5 1 1 877520
#chr start end size gene exon 1 69482 69600 118 OR4F5 1 1 877520 877636 116 SAMD11 8 1 877807 877873 66 SAMD11 9 1 877934 878066 132 SAMD11 10 1 878067 878068 1 SAMD11 10 1 878070 878080 10 SAMD11 10 1 896670 896724 54 KLHL17 2 1 896726 896728 2 KLHL17 2 1 935267 935268 1 HES4 1 1 935271 935357 86 HES4 1 1 955548 955694 146 AGRN 1 1 955720 955758 38 AGRN 1 1 984242 984422 180 AGRN 24 1 984611 984629 18 AGRN 25 1 989928 989936 8 AGRN 35 1 1132946 1133034 88 TTLL10 13 1 1149358 1149397 39 TNFRSF4 1 1 1167654 1167713 59 B3GALT6 1 1 1167733 1167853 120 B3GALT6 1 1 1167878 1167908 30 B3GALT6 1 1 1181889 1182075 186 FAM132A 1 1 1200204 1200215 11 UBE2J2 2 1 1219462 1219466 4 SCNN1D 5 1 1223355 1223358 3 SCNN1D 12 1 1232009 1232018 9 ACAP3 15 1 1244308 1244320 12 PUSL1 2 1 1244321 1244327 6 PUSL1 2 1 1247601 1247603 2 CPSF3L 15 1 1290483 1290485 2 MXRA8 5 1 1290487 1290488 1 MXRA8 5 1 1290492 1290516 24 MXRA8 5 1 1290619 1290631 12 MXRA8 4 1 1291003 1291136 133 MXRA8 3 1 1292056 1292089 33 MXRA8 2 1 1334399 1334405 6 CCNL2 1 1 1355427 1355489 62 LOC441869 2 1 1355659 1355917 258 LOC441869 2 1 1361505 1361521 16 TMEM88B 1 1 1361523 1361525 2 TMEM88B 1 1 1361527 1361528 1 TMEM88B 1 1 1361635 1361636 1 TMEM88B 1 1 1361642 1361726 84 TMEM88B 1 1 1361757 1361761 4 TMEM88B 1 1 1362931 1362956 25 TMEM88B 2 1 1374780 1374838 58 VWA1 3 1 1374841 1374842 1 VWA1 3 1 1374934 1375100 166 VWA1 3 1 1389738 1389747 9 ATAD3C 4 1 1389751 1389816 65 ATAD3C 4 1 1389830 1389849 19 ATAD3C 4 1 1390835 1390837 2 ATAD3C 5 1 1407260 1407331 71 ATAD3B 1 1 1407340 1407474 -

Literature Mining Sustains and Enhances Knowledge Discovery from Omic Studies
LITERATURE MINING SUSTAINS AND ENHANCES KNOWLEDGE DISCOVERY FROM OMIC STUDIES by Rick Matthew Jordan B.S. Biology, University of Pittsburgh, 1996 M.S. Molecular Biology/Biotechnology, East Carolina University, 2001 M.S. Biomedical Informatics, University of Pittsburgh, 2005 Submitted to the Graduate Faculty of School of Medicine in partial fulfillment of the requirements for the degree of Doctor of Philosophy University of Pittsburgh 2016 UNIVERSITY OF PITTSBURGH SCHOOL OF MEDICINE This dissertation was presented by Rick Matthew Jordan It was defended on December 2, 2015 and approved by Shyam Visweswaran, M.D., Ph.D., Associate Professor Rebecca Jacobson, M.D., M.S., Professor Songjian Lu, Ph.D., Assistant Professor Dissertation Advisor: Vanathi Gopalakrishnan, Ph.D., Associate Professor ii Copyright © by Rick Matthew Jordan 2016 iii LITERATURE MINING SUSTAINS AND ENHANCES KNOWLEDGE DISCOVERY FROM OMIC STUDIES Rick Matthew Jordan, M.S. University of Pittsburgh, 2016 Genomic, proteomic and other experimentally generated data from studies of biological systems aiming to discover disease biomarkers are currently analyzed without sufficient supporting evidence from the literature due to complexities associated with automated processing. Extracting prior knowledge about markers associated with biological sample types and disease states from the literature is tedious, and little research has been performed to understand how to use this knowledge to inform the generation of classification models from ‘omic’ data. Using pathway analysis methods to better understand the underlying biology of complex diseases such as breast and lung cancers is state-of-the-art. However, the problem of how to combine literature- mining evidence with pathway analysis evidence is an open problem in biomedical informatics research. -

Human Induced Pluripotent Stem Cell–Derived Podocytes Mature Into Vascularized Glomeruli Upon Experimental Transplantation
BASIC RESEARCH www.jasn.org Human Induced Pluripotent Stem Cell–Derived Podocytes Mature into Vascularized Glomeruli upon Experimental Transplantation † Sazia Sharmin,* Atsuhiro Taguchi,* Yusuke Kaku,* Yasuhiro Yoshimura,* Tomoko Ohmori,* ‡ † ‡ Tetsushi Sakuma, Masashi Mukoyama, Takashi Yamamoto, Hidetake Kurihara,§ and | Ryuichi Nishinakamura* *Department of Kidney Development, Institute of Molecular Embryology and Genetics, and †Department of Nephrology, Faculty of Life Sciences, Kumamoto University, Kumamoto, Japan; ‡Department of Mathematical and Life Sciences, Graduate School of Science, Hiroshima University, Hiroshima, Japan; §Division of Anatomy, Juntendo University School of Medicine, Tokyo, Japan; and |Japan Science and Technology Agency, CREST, Kumamoto, Japan ABSTRACT Glomerular podocytes express proteins, such as nephrin, that constitute the slit diaphragm, thereby contributing to the filtration process in the kidney. Glomerular development has been analyzed mainly in mice, whereas analysis of human kidney development has been minimal because of limited access to embryonic kidneys. We previously reported the induction of three-dimensional primordial glomeruli from human induced pluripotent stem (iPS) cells. Here, using transcription activator–like effector nuclease-mediated homologous recombination, we generated human iPS cell lines that express green fluorescent protein (GFP) in the NPHS1 locus, which encodes nephrin, and we show that GFP expression facilitated accurate visualization of nephrin-positive podocyte formation in -

Termination of RNA Polymerase II Transcription by the 5’-3’ Exonuclease Xrn2
TERMINATION OF RNA POLYMERASE II TRANSCRIPTION BY THE 5’-3’ EXONUCLEASE XRN2 by MICHAEL ANDRES CORTAZAR OSORIO B.S., Universidad del Valle – Colombia, 2011 A thesis submitted to the Faculty of the Graduate School of the University of Colorado in partial fulfillment of the requirements for the degree of Doctor of Philosophy Molecular Biology Program 2018 This thesis for the Doctor of Philosophy degree by Michael Andrés Cortázar Osorio has been approved for the Molecular Biology Program by Mair Churchill, Chair Richard Davis Jay Hesselberth Thomas Blumenthal James Goodrich David Bentley, Advisor Date: Aug 17, 2018 ii Cortázar Osorio, Michael Andrés (Ph.D., Molecular Biology) Termination of RNA polymerase II transcription by the 5’-3’ exonuclease Xrn2 Thesis directed by Professor David L. Bentley ABSTRACT Termination of transcription occurs when RNA polymerase (pol) II dissociates from the DNA template and releases a newly-made mRNA molecule. Interestingly, an active debate fueled by conflicting reports over the last three decades is still open on which of the two main models of termination of RNA polymerase II transcription does in fact operate at 3’ ends of genes. The torpedo model indicates that the 5’-3’ exonuclease Xrn2 targets the nascent transcript for degradation after cleavage at the polyA site and chases pol II for termination. In contrast, the allosteric model asserts that transcription through the polyA signal induces a conformational change of the elongation complex and converts it into a termination-competent complex. In this thesis, I propose a unified allosteric-torpedo mechanism. Consistent with a polyA site-dependent conformational change of the elongation complex, I found that pol II transitions at the polyA site into a mode of slow transcription elongation that is accompanied by loss of Spt5 phosphorylation in the elongation complex.