Assembly and Annotation of an Ashkenazi Human Reference Genome
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Supplemental Note Hominoid Fission of Chromosome 14/15 and Role Of
Supplemental Note Supplemental Note Hominoid fission of chromosome 14/15 and role of segmental duplications Giuliana Giannuzzi, Michele Pazienza, John Huddleston, Francesca Antonacci, Maika Malig, Laura Vives, Evan E. Eichler and Mario Ventura 1. Analysis of the macaque contig spanning the hominoid 14/15 fission site We grouped contig clones based on their FISH pattern on human and macaque chromosomes (Groups F1, F2, F3, G1, G2, G3, and G4). Group F2 clones (Hsa15b- and Hsa15c-positive) showed a single signal on macaque 7q and three signal clusters on human chromosome 15: at the orthologous 15q26, as expected, as well as at 15q11–14 and 15q24–25, which correspond to the actual and ancestral pericentromeric regions, respectively (Ventura et al. 2003). Indeed, this locus contains an LCR15 copy (Pujana et al. 2001) in both the macaque and human genomes. Most BAC clones were one-end anchored in the human genome (chr15:100,028–100,071 kb). In macaque this region experienced a 64 kb duplicative insertion from chromosome 17 (orthologous to human chromosome 13) (Figure 2), with the human configuration (absence of insertion) likely being the ancestral state because it is identical in orangutan and marmoset. Four Hsa15b- positive clones mapped on macaque and human chromosome 19, but neither human nor macaque assemblies report the STS Hsa15b duplicated at this locus. The presence of assembly gaps in macaque may explain why the STS is not annotated in this region. We aligned the sequence of macaque CH250-70H12 (AC187495.2) versus its human orthologous sequence (hg18 chr15:100,039k-100,170k) and found a 12 kb human expansion through tandem duplication of a ~100 bp unit corresponding to a portion of exon 20 of DNM1 (Figure S3). -

Genetic Variation Across the Human Olfactory Receptor Repertoire Alters Odor Perception
bioRxiv preprint doi: https://doi.org/10.1101/212431; this version posted November 1, 2017. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. Genetic variation across the human olfactory receptor repertoire alters odor perception Casey Trimmer1,*, Andreas Keller2, Nicolle R. Murphy1, Lindsey L. Snyder1, Jason R. Willer3, Maira Nagai4,5, Nicholas Katsanis3, Leslie B. Vosshall2,6,7, Hiroaki Matsunami4,8, and Joel D. Mainland1,9 1Monell Chemical Senses Center, Philadelphia, Pennsylvania, USA 2Laboratory of Neurogenetics and Behavior, The Rockefeller University, New York, New York, USA 3Center for Human Disease Modeling, Duke University Medical Center, Durham, North Carolina, USA 4Department of Molecular Genetics and Microbiology, Duke University Medical Center, Durham, North Carolina, USA 5Department of Biochemistry, University of Sao Paulo, Sao Paulo, Brazil 6Howard Hughes Medical Institute, New York, New York, USA 7Kavli Neural Systems Institute, New York, New York, USA 8Department of Neurobiology and Duke Institute for Brain Sciences, Duke University Medical Center, Durham, North Carolina, USA 9Department of Neuroscience, University of Pennsylvania School of Medicine, Philadelphia, Pennsylvania, USA *[email protected] ABSTRACT The human olfactory receptor repertoire is characterized by an abundance of genetic variation that affects receptor response, but the perceptual effects of this variation are unclear. To address this issue, we sequenced the OR repertoire in 332 individuals and examined the relationship between genetic variation and 276 olfactory phenotypes, including the perceived intensity and pleasantness of 68 odorants at two concentrations, detection thresholds of three odorants, and general olfactory acuity. -

(PWP2) Gene Mapping to 21Q22.3 Ronald G
Downloaded from genome.cshlp.org on October 2, 2021 - Published by Cold Spring Harbor Laboratory Press LETTER Isolation and Genomic Structure of a Human Homolog of the Yeast Periodic Tryptophan Protein 2 (PWP2) Gene Mapping to 21q22.3 Ronald G. Lafrenii~re, 1'4 Daniel L. Rochefort, 1 Nathalie Chr~tien, ~ Catherine E. Neville, 2 Robert G. Korneluk, 2 Lin Zuo, 3 Yalin Wei, 3 Jay Lichter, 3 and Guy A. Rouleau ~ 1Centre for Research in Neuroscience, McGill University, and Department of Neurology, Montreal General Hospital Research Institute, Montreal, Canada H3G 1A4; 2DNA Sequencing Core Facility, Canadian Genetic Diseases Network, Children's Hospital of Eastern Ontario, Ottawa, Canada K1H 8L1; 3Sequana Therapeutics, Inc., La Jolla, California 92037 As part of efforts to identify candidate genes for diseases mapping to the 21q22.3 region, we have assembled a 770-kb cosmid and BAC contig containing eight tightly linked markers. These cosmids and BACs were restriction mapped using eight rare cutting enzymes, with the goal of identifying CpG-rich islands. One such island was identified by the clustering of lqotl, Eagl, Sstll, and BssHIl sites, and corresponded to the Nod linking clone LJI04 described previously. A 7.6-kb l-lindlll fragment containing this CpG-rich island was subcloned and partially sequenced. A homology search using the sequence obtained from either side of the Nod site identified an expressed sequence tag with homology to the yeast periodic tryptophan protein 2 (PWP2). Several cDNAs corresponding to the human PWP2 gene were identified and partially sequenced. Northern blot analysis revealed a 3.3-kb transcript that was well expressed in all tissues tested. -

WO 2019/079361 Al 25 April 2019 (25.04.2019) W 1P O PCT
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization I International Bureau (10) International Publication Number (43) International Publication Date WO 2019/079361 Al 25 April 2019 (25.04.2019) W 1P O PCT (51) International Patent Classification: CA, CH, CL, CN, CO, CR, CU, CZ, DE, DJ, DK, DM, DO, C12Q 1/68 (2018.01) A61P 31/18 (2006.01) DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, HN, C12Q 1/70 (2006.01) HR, HU, ID, IL, IN, IR, IS, JO, JP, KE, KG, KH, KN, KP, KR, KW, KZ, LA, LC, LK, LR, LS, LU, LY, MA, MD, ME, (21) International Application Number: MG, MK, MN, MW, MX, MY, MZ, NA, NG, NI, NO, NZ, PCT/US2018/056167 OM, PA, PE, PG, PH, PL, PT, QA, RO, RS, RU, RW, SA, (22) International Filing Date: SC, SD, SE, SG, SK, SL, SM, ST, SV, SY, TH, TJ, TM, TN, 16 October 2018 (16. 10.2018) TR, TT, TZ, UA, UG, US, UZ, VC, VN, ZA, ZM, ZW. (25) Filing Language: English (84) Designated States (unless otherwise indicated, for every kind of regional protection available): ARIPO (BW, GH, (26) Publication Language: English GM, KE, LR, LS, MW, MZ, NA, RW, SD, SL, ST, SZ, TZ, (30) Priority Data: UG, ZM, ZW), Eurasian (AM, AZ, BY, KG, KZ, RU, TJ, 62/573,025 16 October 2017 (16. 10.2017) US TM), European (AL, AT, BE, BG, CH, CY, CZ, DE, DK, EE, ES, FI, FR, GB, GR, HR, HU, ΓΕ , IS, IT, LT, LU, LV, (71) Applicant: MASSACHUSETTS INSTITUTE OF MC, MK, MT, NL, NO, PL, PT, RO, RS, SE, SI, SK, SM, TECHNOLOGY [US/US]; 77 Massachusetts Avenue, TR), OAPI (BF, BJ, CF, CG, CI, CM, GA, GN, GQ, GW, Cambridge, Massachusetts 02139 (US). -

De Novo Genome Assembly and Analysis of Non- Allelic Recombination in Pathogenic Yeast Candida Glabrata
DE NOVO GENOME ASSEMBLY AND ANALYSIS OF NON- ALLELIC RECOMBINATION IN PATHOGENIC YEAST CANDIDA GLABRATA by Zhuwei Xu A dissertation submitted to Johns Hopkins University in conformity with the requirements for the degree of Doctor of Philosophy Baltimore, Maryland November 2019 © 2019 Zhuwei Xu All Rights Reserved Abstract Candida glabrata is an opportunistic pathogen in humans, responsible for approximately 20% of disseminated candidiasis. C. glabrata’s ability to adhere to host tissue is mediated by GPI- anchored cell wall proteins (GPI-CWPs); the corresponding genes contain long tandem repeat regions and form large gene families. These tandem repeats cause mis-assemblies of GPI-CWP genes in C. glabrata genome. Subtelomeres of C. glabrata are particularly rich in GPI-CWP genes and share homology with each other. Consequently, the subtelomeres are mis-assembled in genome sequences assembled from short sequencing reads. In this thesis, we used the long single-molecule real time (SMRT) reads and performed de novo genome assembly of the C. glabrata genome to establish the correct structure of GPI-CWP genes and the subtelomeres. We assembled the genome of six C. glabrata strains: the type strain, CBS138; our lab strain, BG2; four serial clinical isolates, BG3993-96 to assess genome changes during infection. With high quality sequences in hand, we then assess recombinational exchange between GPI-CWP genes by non-allelic mitotic recombination. This question is difficult to address with normal aligners, and we developed a k-mer based method to identify recombination. Our assembly established the correct subtelomere structure of Candida glabrata and provides correct structure of the GPI-CWP gene families. -
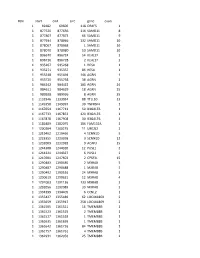
Chr Start End Size Gene Exon 1 69482 69600 118 OR4F5 1 1 877520
#chr start end size gene exon 1 69482 69600 118 OR4F5 1 1 877520 877636 116 SAMD11 8 1 877807 877873 66 SAMD11 9 1 877934 878066 132 SAMD11 10 1 878067 878068 1 SAMD11 10 1 878070 878080 10 SAMD11 10 1 896670 896724 54 KLHL17 2 1 896726 896728 2 KLHL17 2 1 935267 935268 1 HES4 1 1 935271 935357 86 HES4 1 1 955548 955694 146 AGRN 1 1 955720 955758 38 AGRN 1 1 984242 984422 180 AGRN 24 1 984611 984629 18 AGRN 25 1 989928 989936 8 AGRN 35 1 1132946 1133034 88 TTLL10 13 1 1149358 1149397 39 TNFRSF4 1 1 1167654 1167713 59 B3GALT6 1 1 1167733 1167853 120 B3GALT6 1 1 1167878 1167908 30 B3GALT6 1 1 1181889 1182075 186 FAM132A 1 1 1200204 1200215 11 UBE2J2 2 1 1219462 1219466 4 SCNN1D 5 1 1223355 1223358 3 SCNN1D 12 1 1232009 1232018 9 ACAP3 15 1 1244308 1244320 12 PUSL1 2 1 1244321 1244327 6 PUSL1 2 1 1247601 1247603 2 CPSF3L 15 1 1290483 1290485 2 MXRA8 5 1 1290487 1290488 1 MXRA8 5 1 1290492 1290516 24 MXRA8 5 1 1290619 1290631 12 MXRA8 4 1 1291003 1291136 133 MXRA8 3 1 1292056 1292089 33 MXRA8 2 1 1334399 1334405 6 CCNL2 1 1 1355427 1355489 62 LOC441869 2 1 1355659 1355917 258 LOC441869 2 1 1361505 1361521 16 TMEM88B 1 1 1361523 1361525 2 TMEM88B 1 1 1361527 1361528 1 TMEM88B 1 1 1361635 1361636 1 TMEM88B 1 1 1361642 1361726 84 TMEM88B 1 1 1361757 1361761 4 TMEM88B 1 1 1362931 1362956 25 TMEM88B 2 1 1374780 1374838 58 VWA1 3 1 1374841 1374842 1 VWA1 3 1 1374934 1375100 166 VWA1 3 1 1389738 1389747 9 ATAD3C 4 1 1389751 1389816 65 ATAD3C 4 1 1389830 1389849 19 ATAD3C 4 1 1390835 1390837 2 ATAD3C 5 1 1407260 1407331 71 ATAD3B 1 1 1407340 1407474 -

Investigation of RNA Binding Proteins Regulated by Mtor
Investigation of RNA binding proteins regulated by mTOR Thesis submitted to the University of Leicester for the degree of Doctor of Philosophy Katherine Morris BSc (University of Leicester) March 2017 1 Investigation of RNA binding proteins regulated by mTOR Katherine Morris, MRC Toxicology Unit, University of Leicester, Leicester, LE1 9HN The mammalian target of rapamycin (mTOR) is a serine/threonine protein kinase which plays a key role in the transduction of cellular energy signals, in order to coordinate and regulate a wide number of processes including cell growth and proliferation via control of protein synthesis and protein degradation. For a number of human diseases where mTOR signalling is dysregulated, including cancer, the clinical relevance of mTOR inhibitors is clear. However, understanding of the mechanisms by which mTOR controls gene expression is incomplete, with implications for adverse toxicological effects of mTOR inhibitors on clinical outcomes. mTOR has been shown to regulate 5’ TOP mRNA expression, though the exact mechanism remains unclear. It has been postulated that this may involve an intermediary factor such as an RNA binding protein, which acts downstream of mTOR signalling to bind and regulate translation or stability of specific messages. This thesis aimed to address this question through the use of whole cell RNA binding protein capture using oligo‐d(T) affinity isolation and subsequent proteomic analysis, and identify RNA binding proteins with differential binding activity following mTOR inhibition. Following validation of 4 identified mTOR‐dependent RNA binding proteins, characterisation of their specific functions with respect to growth and survival was conducted through depletion studies, identifying a promising candidate for further work; LARP1. -

Hippo and Sonic Hedgehog Signalling Pathway Modulation of Human Urothelial Tissue Homeostasis
Hippo and Sonic Hedgehog signalling pathway modulation of human urothelial tissue homeostasis Thomas Crighton PhD University of York Department of Biology November 2020 Abstract The urinary tract is lined by a barrier-forming, mitotically-quiescent urothelium, which retains the ability to regenerate following injury. Regulation of tissue homeostasis by Hippo and Sonic Hedgehog signalling has previously been implicated in various mammalian epithelia, but limited evidence exists as to their role in adult human urothelial physiology. Focussing on the Hippo pathway, the aims of this thesis were to characterise expression of said pathways in urothelium, determine what role the pathways have in regulating urothelial phenotype, and investigate whether the pathways are implicated in muscle-invasive bladder cancer (MIBC). These aims were assessed using a cell culture paradigm of Normal Human Urothelial (NHU) cells that can be manipulated in vitro to represent different differentiated phenotypes, alongside MIBC cell lines and The Cancer Genome Atlas resource. Transcriptomic analysis of NHU cells identified a significant induction of VGLL1, a poorly understood regulator of Hippo signalling, in differentiated cells. Activation of upstream transcription factors PPARγ and GATA3 and/or blockade of active EGFR/RAS/RAF/MEK/ERK signalling were identified as mechanisms which induce VGLL1 expression in NHU cells. Ectopic overexpression of VGLL1 in undifferentiated NHU cells and MIBC cell line T24 resulted in significantly reduced proliferation. Conversely, knockdown of VGLL1 in differentiated NHU cells significantly reduced barrier tightness in an unwounded state, while inhibiting regeneration and increasing cell cycle activation in scratch-wounded cultures. A signalling pathway previously observed to be inhibited by VGLL1 function, YAP/TAZ, was unaffected by VGLL1 manipulation. -

Role and Regulation of the P53-Homolog P73 in the Transformation of Normal Human Fibroblasts
Role and regulation of the p53-homolog p73 in the transformation of normal human fibroblasts Dissertation zur Erlangung des naturwissenschaftlichen Doktorgrades der Bayerischen Julius-Maximilians-Universität Würzburg vorgelegt von Lars Hofmann aus Aschaffenburg Würzburg 2007 Eingereicht am Mitglieder der Promotionskommission: Vorsitzender: Prof. Dr. Dr. Martin J. Müller Gutachter: Prof. Dr. Michael P. Schön Gutachter : Prof. Dr. Georg Krohne Tag des Promotionskolloquiums: Doktorurkunde ausgehändigt am Erklärung Hiermit erkläre ich, dass ich die vorliegende Arbeit selbständig angefertigt und keine anderen als die angegebenen Hilfsmittel und Quellen verwendet habe. Diese Arbeit wurde weder in gleicher noch in ähnlicher Form in einem anderen Prüfungsverfahren vorgelegt. Ich habe früher, außer den mit dem Zulassungsgesuch urkundlichen Graden, keine weiteren akademischen Grade erworben und zu erwerben gesucht. Würzburg, Lars Hofmann Content SUMMARY ................................................................................................................ IV ZUSAMMENFASSUNG ............................................................................................. V 1. INTRODUCTION ................................................................................................. 1 1.1. Molecular basics of cancer .......................................................................................... 1 1.2. Early research on tumorigenesis ................................................................................. 3 1.3. Developing -

The Genetics of Bipolar Disorder
Molecular Psychiatry (2008) 13, 742–771 & 2008 Nature Publishing Group All rights reserved 1359-4184/08 $30.00 www.nature.com/mp FEATURE REVIEW The genetics of bipolar disorder: genome ‘hot regions,’ genes, new potential candidates and future directions A Serretti and L Mandelli Institute of Psychiatry, University of Bologna, Bologna, Italy Bipolar disorder (BP) is a complex disorder caused by a number of liability genes interacting with the environment. In recent years, a large number of linkage and association studies have been conducted producing an extremely large number of findings often not replicated or partially replicated. Further, results from linkage and association studies are not always easily comparable. Unfortunately, at present a comprehensive coverage of available evidence is still lacking. In the present paper, we summarized results obtained from both linkage and association studies in BP. Further, we indicated new potential interesting genes, located in genome ‘hot regions’ for BP and being expressed in the brain. We reviewed published studies on the subject till December 2007. We precisely localized regions where positive linkage has been found, by the NCBI Map viewer (http://www.ncbi.nlm.nih.gov/mapview/); further, we identified genes located in interesting areas and expressed in the brain, by the Entrez gene, Unigene databases (http://www.ncbi.nlm.nih.gov/entrez/) and Human Protein Reference Database (http://www.hprd.org); these genes could be of interest in future investigations. The review of association studies gave interesting results, as a number of genes seem to be definitively involved in BP, such as SLC6A4, TPH2, DRD4, SLC6A3, DAOA, DTNBP1, NRG1, DISC1 and BDNF. -

WO 2012/174282 A2 20 December 2012 (20.12.2012) P O P C T
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date WO 2012/174282 A2 20 December 2012 (20.12.2012) P O P C T (51) International Patent Classification: David [US/US]; 13539 N . 95th Way, Scottsdale, AZ C12Q 1/68 (2006.01) 85260 (US). (21) International Application Number: (74) Agent: AKHAVAN, Ramin; Caris Science, Inc., 6655 N . PCT/US20 12/0425 19 Macarthur Blvd., Irving, TX 75039 (US). (22) International Filing Date: (81) Designated States (unless otherwise indicated, for every 14 June 2012 (14.06.2012) kind of national protection available): AE, AG, AL, AM, AO, AT, AU, AZ, BA, BB, BG, BH, BR, BW, BY, BZ, English (25) Filing Language: CA, CH, CL, CN, CO, CR, CU, CZ, DE, DK, DM, DO, Publication Language: English DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, HN, HR, HU, ID, IL, IN, IS, JP, KE, KG, KM, KN, KP, KR, (30) Priority Data: KZ, LA, LC, LK, LR, LS, LT, LU, LY, MA, MD, ME, 61/497,895 16 June 201 1 (16.06.201 1) US MG, MK, MN, MW, MX, MY, MZ, NA, NG, NI, NO, NZ, 61/499,138 20 June 201 1 (20.06.201 1) US OM, PE, PG, PH, PL, PT, QA, RO, RS, RU, RW, SC, SD, 61/501,680 27 June 201 1 (27.06.201 1) u s SE, SG, SK, SL, SM, ST, SV, SY, TH, TJ, TM, TN, TR, 61/506,019 8 July 201 1(08.07.201 1) u s TT, TZ, UA, UG, US, UZ, VC, VN, ZA, ZM, ZW. -

Down Syndrome Congenital Heart Disease: a Narrowed Region and a Candidate Gene Gillian M
March/April 2001 ⅐ Vol. 3 ⅐ No. 2 article Down syndrome congenital heart disease: A narrowed region and a candidate gene Gillian M. Barlow, PhD1, Xiao-Ning Chen, MD1, Zheng Y. Shi, BS1, Gary E. Lyons, PhD2, David M. Kurnit, MD, PhD3, Livija Celle, MS4, Nancy B. Spinner, PhD4, Elaine Zackai, MD4, Mark J. Pettenati, PhD5, Alexander J. Van Riper, MS6, Michael J. Vekemans, MD7, Corey H. Mjaatvedt, PhD8, and Julie R. Korenberg, PhD, MD1 Purpose: Down syndrome (DS) is a major cause of congenital heart disease (CHD) and the most frequent known cause of atrioventricular septal defects (AVSDs). Molecular studies of rare individuals with CHD and partial duplications of chromosome 21 established a candidate region that included D21S55 through the telomere. We now report human molecular and cardiac data that narrow the DS-CHD region, excluding two candidate regions, and propose DSCAM (Down syndrome cell adhesion molecule) as a candidate gene. Methods: A panel of 19 individuals with partial trisomy 21 was evaluated using quantitative Southern blot dosage analysis and fluorescence in situ hybridization (FISH) with subsets of 32 BACs spanning the region defined by D21S16 (21q11.2) through the telomere. These BACs span the molecular markers D21S55, ERG, ETS2, MX1/2, collagen XVIII and collagen VI A1/A2. Fourteen individuals are duplicated for the candidate region, of whom eight (57%) have the characteristic spectrum of DS-CHD. Results: Combining the results from these eight individuals suggests the candidate region for DS-CHD is demarcated by D21S3 (defined by ventricular septal defect), through PFKL (defined by tetralogy of Fallot). Conclusions: These data suggest that the presence of three copies of gene(s) from the region is sufficient for the production of subsets of DS-CHD.