Functional Analysis of Human Hub Proteins and Their Interactors Involved in the Intrinsic Disorder-Enriched Interactions
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Regulates Cellular Telomerase Activity by Methylation of TERT Promoter
www.impactjournals.com/oncotarget/ Oncotarget, 2017, Vol. 8, (No. 5), pp: 7977-7988 Research Paper Tianshengyuan-1 (TSY-1) regulates cellular Telomerase activity by methylation of TERT promoter Weibo Yu1, Xiaotian Qin2, Yusheng Jin1, Yawei Li2, Chintda Santiskulvong3, Victor Vu1, Gang Zeng4,5, Zuofeng Zhang6, Michelle Chow1, Jianyu Rao1,5 1Department of Pathology and Laboratory Medicine, David Geffen School of Medicine, University of California at Los Angeles, Los Angeles, CA, USA 2Beijing Boyuantaihe Biological Technology Co., Ltd., Beijing, China 3Genomics Core, Cedars-Sinai Medical Center, Los Angeles, CA, USA 4Department of Urology, David Geffen School of Medicine, University of California at Los Angeles, Los Angeles, CA, USA 5Jonsson Comprehensive Cancer Center, University of California at Los Angeles, Los Angeles, CA, USA 6Department of Epidemiology, School of Public Health, University of California at Los Angeles, Los Angeles, CA, USA Correspondence to: Jianyu Rao, email: [email protected] Keywords: TSY-1, hematopoietic cells, Telomerase, TERT, methylation Received: September 08, 2016 Accepted: November 24, 2016 Published: December 15, 2016 ABSTRACT Telomere and Telomerase have recently been explored as anti-aging and anti- cancer drug targets with only limited success. Previously we showed that the Chinese herbal medicine Tianshengyuan-1 (TSY-1), an agent used to treat bone marrow deficiency, has a profound effect on stimulating Telomerase activity in hematopoietic cells. Here, the mechanism of TSY-1 on cellular Telomerase activity was further investigated using HL60, a promyelocytic leukemia cell line, normal peripheral blood mononuclear cells, and CD34+ hematopoietic stem cells derived from umbilical cord blood. TSY-1 increases Telomerase activity in normal peripheral blood mononuclear cells and CD34+ hematopoietic stem cells with innately low Telomerase activity but decreases Telomerase activity in HL60 cells with high intrinsic Telomerase activity, both in a dose-response manner. -
![Downloaded from [266]](https://docslib.b-cdn.net/cover/7352/downloaded-from-266-347352.webp)
Downloaded from [266]
Patterns of DNA methylation on the human X chromosome and use in analyzing X-chromosome inactivation by Allison Marie Cotton B.Sc., The University of Guelph, 2005 A THESIS SUBMITTED IN PARTIAL FULFILLMENT OF THE REQUIREMENTS FOR THE DEGREE OF DOCTOR OF PHILOSOPHY in The Faculty of Graduate Studies (Medical Genetics) THE UNIVERSITY OF BRITISH COLUMBIA (Vancouver) January 2012 © Allison Marie Cotton, 2012 Abstract The process of X-chromosome inactivation achieves dosage compensation between mammalian males and females. In females one X chromosome is transcriptionally silenced through a variety of epigenetic modifications including DNA methylation. Most X-linked genes are subject to X-chromosome inactivation and only expressed from the active X chromosome. On the inactive X chromosome, the CpG island promoters of genes subject to X-chromosome inactivation are methylated in their promoter regions, while genes which escape from X- chromosome inactivation have unmethylated CpG island promoters on both the active and inactive X chromosomes. The first objective of this thesis was to determine if the DNA methylation of CpG island promoters could be used to accurately predict X chromosome inactivation status. The second objective was to use DNA methylation to predict X-chromosome inactivation status in a variety of tissues. A comparison of blood, muscle, kidney and neural tissues revealed tissue-specific X-chromosome inactivation, in which 12% of genes escaped from X-chromosome inactivation in some, but not all, tissues. X-linked DNA methylation analysis of placental tissues predicted four times higher escape from X-chromosome inactivation than in any other tissue. Despite the hypomethylation of repetitive elements on both the X chromosome and the autosomes, no changes were detected in the frequency or intensity of placental Cot-1 holes. -

Producing T Cells
Lnk/Sh2b3 Controls the Production and Function of Dendritic Cells and Regulates the Induction of IFN- −γ Producing T Cells This information is current as Taizo Mori, Yukiko Iwasaki, Yoichi Seki, Masanori Iseki, of September 28, 2021. Hiroko Katayama, Kazuhiko Yamamoto, Kiyoshi Takatsu and Satoshi Takaki J Immunol published online 14 July 2014 http://www.jimmunol.org/content/early/2014/07/13/jimmun ol.1303243 Downloaded from Supplementary http://www.jimmunol.org/content/suppl/2014/07/14/jimmunol.130324 Material 3.DCSupplemental http://www.jimmunol.org/ Why The JI? Submit online. • Rapid Reviews! 30 days* from submission to initial decision • No Triage! Every submission reviewed by practicing scientists • Fast Publication! 4 weeks from acceptance to publication by guest on September 28, 2021 *average Subscription Information about subscribing to The Journal of Immunology is online at: http://jimmunol.org/subscription Permissions Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Email Alerts Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts The Journal of Immunology is published twice each month by The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2014 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. Published July 14, 2014, doi:10.4049/jimmunol.1303243 The Journal of Immunology Lnk/Sh2b3 Controls the Production and Function of Dendritic Cells and Regulates the Induction of IFN-g–Producing T Cells Taizo Mori,*,1 Yukiko Iwasaki,*,†,1 Yoichi Seki,* Masanori Iseki,* Hiroko Katayama,* Kazuhiko Yamamoto,† Kiyoshi Takatsu,‡,x and Satoshi Takaki* Dendritic cells (DCs) are proficient APCs that play crucial roles in the immune responses to various Ags and pathogens and polarize Th cell immune responses. -

Genetic and Genomic Analysis of Hyperlipidemia, Obesity and Diabetes Using (C57BL/6J × TALLYHO/Jngj) F2 Mice
University of Tennessee, Knoxville TRACE: Tennessee Research and Creative Exchange Nutrition Publications and Other Works Nutrition 12-19-2010 Genetic and genomic analysis of hyperlipidemia, obesity and diabetes using (C57BL/6J × TALLYHO/JngJ) F2 mice Taryn P. Stewart Marshall University Hyoung Y. Kim University of Tennessee - Knoxville, [email protected] Arnold M. Saxton University of Tennessee - Knoxville, [email protected] Jung H. Kim Marshall University Follow this and additional works at: https://trace.tennessee.edu/utk_nutrpubs Part of the Animal Sciences Commons, and the Nutrition Commons Recommended Citation BMC Genomics 2010, 11:713 doi:10.1186/1471-2164-11-713 This Article is brought to you for free and open access by the Nutrition at TRACE: Tennessee Research and Creative Exchange. It has been accepted for inclusion in Nutrition Publications and Other Works by an authorized administrator of TRACE: Tennessee Research and Creative Exchange. For more information, please contact [email protected]. Stewart et al. BMC Genomics 2010, 11:713 http://www.biomedcentral.com/1471-2164/11/713 RESEARCH ARTICLE Open Access Genetic and genomic analysis of hyperlipidemia, obesity and diabetes using (C57BL/6J × TALLYHO/JngJ) F2 mice Taryn P Stewart1, Hyoung Yon Kim2, Arnold M Saxton3, Jung Han Kim1* Abstract Background: Type 2 diabetes (T2D) is the most common form of diabetes in humans and is closely associated with dyslipidemia and obesity that magnifies the mortality and morbidity related to T2D. The genetic contribution to human T2D and related metabolic disorders is evident, and mostly follows polygenic inheritance. The TALLYHO/ JngJ (TH) mice are a polygenic model for T2D characterized by obesity, hyperinsulinemia, impaired glucose uptake and tolerance, hyperlipidemia, and hyperglycemia. -
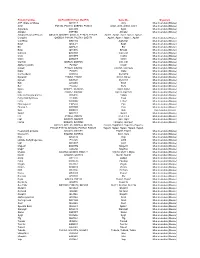
Gene Ids Organism ATP Citrate Synthase Q3V117 Acly
Protein Families UniProtKB ID (from MudPIT) Gene IDs Organism ATP citrate synthase Q3V117 Acly Mus musculus (Mouse) Actin P68134, P60710, Q8BFZ3, P68033 Acta1, Actb, Actbl2, Actc1 Mus musculus (Mouse) Argonaute Q8CJG0 Ago2 Mus musculus (Mouse) Ahnak2 E9PYB0 Ahnak2 Mus musculus (Mouse) Adaptor Related Protein Q8CC13, Q8CBB7, Q3UHJ0, P17426, P17427, Ap1b1, Ap1g1, Aak1, Ap2a1, Ap2a2, Complex Q9DBG3, P84091, P62743, Q9Z1T1 Ap2b1, Ap2m1, Ap2s1 , Ap3b1 Mus musculus (Mouse) V-ATPase Q9Z1G4 Atp6v0a1 Mus musculus (Mouse) Bag3 Q9JLV1 Bag3 Mus musculus (Mouse) Bcr Q6PAJ1 Bcr Mus musculus (Mouse) Bmp Q91Z96 Bmp2k Mus musculus (Mouse) Calcoco Q8CGU1 Calcoco1 Mus musculus (Mouse) Ccdc D3YZP9 Ccdc6 Mus musculus (Mouse) Clint1 Q5SUH7 Clint1 Mus musculus (Mouse) Clathrin Q6IRU5, Q68FD5 Cltb, Cltc Mus musculus (Mouse) Alpha-crystallin P23927 Cryab Mus musculus (Mouse) Casein P19228, Q02862 Csn1s1, Csn1s2a Mus musculus (Mouse) Dab2 P98078 Dab2 Mus musculus (Mouse) Connecdenn Q8K382 Dennd1a Mus musculus (Mouse) Dynamin P39053, P39054 Dnm1, Dnm2 Mus musculus (Mouse) Dynein Q9JHU4 Dync1h1 Mus musculus (Mouse) Edc Q3UJB9 Edc4 Mus musculus (Mouse) Eef P58252 Eef2 Mus musculus (Mouse) Epsin Q80VP1, Q5NCM5 Epn1, Epn2 Mus musculus (Mouse) Eps P42567, Q60902 Eps15, Eps15l1 Mus musculus (Mouse) Fatty acid binding protein Q05816 Fabp5 Mus musculus (Mouse) Fatty Acid Synthase P19096 Fasn Mus musculus (Mouse) Fcho Q3UQN2 Fcho2 Mus musculus (Mouse) Fibrinogen A E9PV24 Fga Mus musculus (Mouse) Filamin A Q8BTM8 Flna Mus musculus (Mouse) Gak Q99KY4 Gak Mus musculus (Mouse) -

Comprehensive Array CGH of Normal Karyotype Myelodysplastic
Leukemia (2011) 25, 387–399 & 2011 Macmillan Publishers Limited All rights reserved 0887-6924/11 www.nature.com/leu LEADING ARTICLE Comprehensive array CGH of normal karyotype myelodysplastic syndromes reveals hidden recurrent and individual genomic copy number alterations with prognostic relevance A Thiel1, M Beier1, D Ingenhag1, K Servan1, M Hein1, V Moeller1, B Betz1, B Hildebrandt1, C Evers1,3, U Germing2 and B Royer-Pokora1 1Institute of Human Genetics and Anthropology, Medical Faculty, Heinrich Heine University, Duesseldorf, Germany and 2Department of Hematology, Oncology and Clinical Immunology, Heinrich Heine University, Duesseldorf, Germany About 40% of patients with myelodysplastic syndromes (MDSs) 40–50% of MDS cases have a normal karyotype. MDS patients present with a normal karyotype, and they are facing different with a normal karyotype and low-risk clinical parameters are courses of disease. To advance the biological understanding often assigned into the IPSS low and intermediate-1 risk groups. and to find molecular prognostic markers, we performed a high- resolution oligonucleotide array study of 107 MDS patients In the absence of genetic or biological markers, prognostic (French American British) with a normal karyotype and clinical stratification of these patients is difficult. To better prognosticate follow-up through the Duesseldorf MDS registry. Recurrent these patients, new parameters to identify patients at higher risk hidden deletions overlapping with known cytogenetic aberra- are urgently needed. With the more recently introduced modern tions or sites of known tumor-associated genes were identi- technologies of whole-genome-wide surveys of genetic aberra- fied in 4q24 (TET2, 2x), 5q31.2 (2x), 7q22.1 (3x) and 21q22.12 tions, it is hoped that more insights into the biology of disease (RUNX1, 2x). -

Supplementary Materials
Supplementary materials Supplementary Table S1: MGNC compound library Ingredien Molecule Caco- Mol ID MW AlogP OB (%) BBB DL FASA- HL t Name Name 2 shengdi MOL012254 campesterol 400.8 7.63 37.58 1.34 0.98 0.7 0.21 20.2 shengdi MOL000519 coniferin 314.4 3.16 31.11 0.42 -0.2 0.3 0.27 74.6 beta- shengdi MOL000359 414.8 8.08 36.91 1.32 0.99 0.8 0.23 20.2 sitosterol pachymic shengdi MOL000289 528.9 6.54 33.63 0.1 -0.6 0.8 0 9.27 acid Poricoic acid shengdi MOL000291 484.7 5.64 30.52 -0.08 -0.9 0.8 0 8.67 B Chrysanthem shengdi MOL004492 585 8.24 38.72 0.51 -1 0.6 0.3 17.5 axanthin 20- shengdi MOL011455 Hexadecano 418.6 1.91 32.7 -0.24 -0.4 0.7 0.29 104 ylingenol huanglian MOL001454 berberine 336.4 3.45 36.86 1.24 0.57 0.8 0.19 6.57 huanglian MOL013352 Obacunone 454.6 2.68 43.29 0.01 -0.4 0.8 0.31 -13 huanglian MOL002894 berberrubine 322.4 3.2 35.74 1.07 0.17 0.7 0.24 6.46 huanglian MOL002897 epiberberine 336.4 3.45 43.09 1.17 0.4 0.8 0.19 6.1 huanglian MOL002903 (R)-Canadine 339.4 3.4 55.37 1.04 0.57 0.8 0.2 6.41 huanglian MOL002904 Berlambine 351.4 2.49 36.68 0.97 0.17 0.8 0.28 7.33 Corchorosid huanglian MOL002907 404.6 1.34 105 -0.91 -1.3 0.8 0.29 6.68 e A_qt Magnogrand huanglian MOL000622 266.4 1.18 63.71 0.02 -0.2 0.2 0.3 3.17 iolide huanglian MOL000762 Palmidin A 510.5 4.52 35.36 -0.38 -1.5 0.7 0.39 33.2 huanglian MOL000785 palmatine 352.4 3.65 64.6 1.33 0.37 0.7 0.13 2.25 huanglian MOL000098 quercetin 302.3 1.5 46.43 0.05 -0.8 0.3 0.38 14.4 huanglian MOL001458 coptisine 320.3 3.25 30.67 1.21 0.32 0.9 0.26 9.33 huanglian MOL002668 Worenine -

The Bacterial Proteasome at the Core of Diverse Degradation Pathways
Research Collection Review Article The Bacterial Proteasome at the Core of Diverse Degradation Pathways Author(s): Müller, Andreas U.; Weber-Ban, Eilika Publication Date: 2019-04-09 Permanent Link: https://doi.org/10.3929/ethz-b-000342497 Originally published in: Frontiers in Molecular Biosciences 6, http://doi.org/10.3389/fmolb.2019.00023 Rights / License: Creative Commons Attribution 4.0 International This page was generated automatically upon download from the ETH Zurich Research Collection. For more information please consult the Terms of use. ETH Library MINI REVIEW published: 09 April 2019 doi: 10.3389/fmolb.2019.00023 The Bacterial Proteasome at the Core of Diverse Degradation Pathways Andreas U. Müller and Eilika Weber-Ban* Department of Biology, Institute of Molecular Biology and Biophysics, ETH Zurich, Zurich, Switzerland Proteasomal protein degradation exists in mycobacteria and other actinobacteria, and expands their repertoire of compartmentalizing protein degradation pathways beyond the usual bacterial types. A product of horizontal gene transfer, bacterial proteasomes have evolved to support the organism’s survival under challenging environmental conditions like nutrient starvation and physical or chemical stresses. Like the eukaryotic 20S proteasome, the bacterial core particle is gated and must associate with a regulator complex to form a fully active protease capable of recruiting and internalizing substrate proteins. By association with diverse regulator complexes that employ different recruitment strategies, the bacterial 20S core particle is able to act in different cellular degradation pathways. In association with the mycobacterial proteasomal ATPase Mpa, the proteasome degrades substrates post-translationally modified with prokaryotic, ubiquitin-like protein Pup in a process called pupylation. -

Characterization of the Macrophage Transcriptome in Glomerulonephritis-Susceptible and -Resistant Rat Strains
Genes and Immunity (2011) 12, 78–89 & 2011 Macmillan Publishers Limited All rights reserved 1466-4879/11 www.nature.com/gene ORIGINAL ARTICLE Characterization of the macrophage transcriptome in glomerulonephritis-susceptible and -resistant rat strains K Maratou1, J Behmoaras2, C Fewings1, P Srivastava1, Z D’Souza1, J Smith3, L Game4, T Cook2 and T Aitman1 1Physiological Genomics and Medicine Group, MRC Clinical Sciences Centre, Imperial College London, London, UK; 2Centre for Complement and Inflammation Research, Imperial College London, London, UK; 3Renal Section, Imperial College London, London, UK and 4Genomics Laboratory, MRC Clinical Sciences Centre, London, UK Crescentic glomerulonephritis (CRGN) is a major cause of rapidly progressive renal failure for which the underlying genetic basis is unknown. Wistar–Kyoto (WKY) rats show marked susceptibility to CRGN, whereas Lewis rats are resistant. Glomerular injury and crescent formation are macrophage dependent and mainly explained by seven quantitative trait loci (Crgn1–7). Here, we used microarray analysis in basal and lipopolysaccharide (LPS)-stimulated macrophages to identify genes that reside on pathways predisposing WKY rats to CRGN. We detected 97 novel positional candidates for the uncharacterized Crgn3–7. We identified 10 additional secondary effector genes with profound differences in expression between the two strains (45-fold change, o1% false discovery rate) for basal and LPS-stimulated macrophages. Moreover, we identified eight genes with differentially expressed alternatively spliced isoforms, by using an in-depth analysis at the probe level that allowed us to discard false positives owing to polymorphisms between the two rat strains. Pathway analysis identified several common linked pathways, enriched for differentially expressed genes, which affect macrophage activation. -

Crucial Role of the SH2B1 PH Domain for the Control of Energy Balance
Diabetes Page 2 of 46 1 Crucial Role of the SH2B1 PH Domain for the Control of 2 Energy Balance 3 4 Anabel Floresa, Lawrence S. Argetsingerb+, Lukas K. J. Stadlerc+, Alvaro E. Malagab, Paul B. 5 Vanderb, Lauren C. DeSantisb, Ray M. Joea,b, Joel M. Clineb, Julia M. Keoghc, Elana Henningc, 6 Ines Barrosod, Edson Mendes de Oliveirac, Gowri Chandrashekarb, Erik S. Clutterb, Yixin Hub, 7 Jeanne Stuckeyf, I. Sadaf Farooqic, Martin G. Myers Jr. a,b,e, Christin Carter-Sua,b,e, g * 8 aCell and Molecular Biology Graduate Program, University of Michigan, Ann Arbor, MI 48109, USA 9 bDepartment of Molecular and Integrative Physiology, University of Michigan, Ann Arbor, MI 48109, USA 10 cUniversity of Cambridge Metabolic Research Laboratories and NIHR Cambridge Biomedical Research Centre, 11 Wellcome Trust-MRC Institute of Metabolic Science, Addenbrooke's Hospital, Cambridge, UK 12 dMRC Epidemiology Unit, Wellcome Trust-MRC Institute of Metabolic Science, Addenbrooke's Hospital, 13 Cambridge, UK 14 eDepartment of Internal Medicine, University of Michigan, Ann Arbor, MI 48109, USA 15 fLife Sciences Institute and Departments of Biological Chemistry and Biophysics, University of Michigan, Ann Arbor, 16 MI 48109, USA 17 +Authors contributed equally to this work 18 gLead contact 19 *Correspondence: [email protected] 20 21 Running title: Role of SH2B1 PH Domain in Energy Balance 22 23 24 25 26 27 28 1 Diabetes Publish Ahead of Print, published online August 22, 2019 Page 3 of 46 Diabetes 29 30 Abstract 31 Disruption of the adaptor protein SH2B1 is associated with severe obesity, insulin resistance and 32 neurobehavioral abnormalities in mice and humans. -

Insight Into the Brain-Specific Alpha Isoform of the Scaffold Protein SH2B1 and Its Rare Obesity-Associated Variants
Insight into the Brain-Specific Alpha Isoform of the Scaffold Protein SH2B1 and its Rare Obesity-Associated Variants By Ray Morris Joe A dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy (Cellular and Molecular Biology) in the University of Michigan 2017 Doctoral Committee: Professor Christin Carter-Su, Chair Professor Ken Inoki Professor Benjamin L. Margolis Professor Donna M. Martin Professor Martin G. Myers © Ray Morris Joe ORCID: 0000-0001-7716-2874 [email protected] All rights reserved 2017 Acknowledgements I’d like to thank my mentor, Christin Carter-Su, for providing me a second opportunity and taking a chance on me after my leave of absence without hesitation. It is an honor to have a mentor guide me through the many challenges I have faced as a graduate student. She has taught me to find passion in my profession, and to strive to push through obstacles and find ways to reach my goals. In addition, she has given me insight to find a balance between work and life by giving me freedom to independently perform my research as well as stories on family and child-rearing. Throughout the many countless days and nights writing grants, manuscripts, and analyzing/interpreting data, it was wonderful to share our past cultural identities with one another. I look forward to having Christy as a mentor, colleague, and friend for all my future endeavors I have after I leave her laboratory. I want to thank my thesis committee, Ken Inoki, Ben Margolis, Donna Martin, and Martin Myers for the numerous insights for my scientific training. -

Interactome of Mirnas and Transcriptome of Human Umbilical Cord Endothelial Cells Exposed to Short-Term Simulated Microgravity
www.nature.com/npjmgrav ARTICLE OPEN Interactome of miRNAs and transcriptome of human umbilical cord endothelial cells exposed to short-term simulated microgravity Dharanibalan Kasiviswanathan1,2,4, Rajadurai Chinnasamy Perumal 3,4, Srinivasan Bhuvaneswari1,2, Pavitra Kumar1, ✉ Lakshmikirupa Sundaresan 1,2, Manuel Philip 3, Sajesh Puthenpurackal Krishnankutty3 and Suvro Chatterjee1,2 Adaptation of humans in low gravity conditions is a matter of utmost importance when efforts are on to a gigantic leap in human space expeditions for tourism and formation of space colonies. In this connection, cardiovascular adaptation in low gravity is a critical component of human space exploration. Deep high-throughput sequencing approach allowed us to analyze the miRNA and mRNA expression profiles in human umbilical cord vein endothelial cells (HUVEC), cultured under gravity (G), and stimulated microgravity (MG) achieved with a clinostat. The present study identified totally 1870 miRNAs differentially expressed in HUVEC under MG condition when compared to the cells subjected to unitary G conditions. The functional association of identified miRNAs targeting specific mRNAs revealed that miRNAs, hsa-mir-496, hsa-mir-151a, hsa-miR-296-3p, hsa-mir-148a, hsa-miR-365b-5p, hsa- miR-3687, hsa-mir-454, hsa-miR-155-5p, and hsa-miR-145-5p differentially regulated the genes involved in cell adhesion, angiogenesis, cell cycle, JAK-STAT signaling, MAPK signaling, nitric oxide signaling, VEGF signaling, and wound healing pathways. Further, the q-PCR based experimental studies of upregulated and downregulated miRNA and mRNAs demonstrate that the above 1234567890():,; reported miRNAs influence the cell proliferation and vascular functions of the HUVEC in MG conditions effectively.