Resolving the 3D Landscape of Transcription-Linked Mammalian Chromatin Folding
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Nuclear Receptor Binding Protein 1 Regulates Intestinal Progenitor Cell Homeostasis and Tumour Formation
Edinburgh Research Explorer Nuclear receptor binding protein 1 regulates intestinal progenitor cell homeostasis and tumour formation Citation for published version: Wilson, CH, Crombie, C, van der Weyden, L, Poulogiannis, G, Rust, AG, Pardo, M, Gracia, T, Yu, L, Choudhary, J, Poulin, GB, McIntyre, RE, Winton, DJ, March, HN, Arends, MJ, Fraser, AG & Adams, DJ 2012, 'Nuclear receptor binding protein 1 regulates intestinal progenitor cell homeostasis and tumour formation', EMBO Journal, vol. 31, no. 11, pp. 2486-97. https://doi.org/10.1038/emboj.2012.91 Digital Object Identifier (DOI): 10.1038/emboj.2012.91 Link: Link to publication record in Edinburgh Research Explorer Document Version: Publisher's PDF, also known as Version of record Published In: EMBO Journal Publisher Rights Statement: This is an openaccess article distributed under the terms of the Creative Commons Attribution License, which permits distribution, and reproduction in any medium, provided the original author and source are credited. This license does not permit commercial exploitation without specific permission. General rights Copyright for the publications made accessible via the Edinburgh Research Explorer is retained by the author(s) and / or other copyright owners and it is a condition of accessing these publications that users recognise and abide by the legal requirements associated with these rights. Take down policy The University of Edinburgh has made every reasonable effort to ensure that Edinburgh Research Explorer content complies with UK legislation. If you believe that the public display of this file breaches copyright please contact [email protected] providing details, and we will remove access to the work immediately and investigate your claim. -

4-6 Weeks Old Female C57BL/6 Mice Obtained from Jackson Labs Were Used for Cell Isolation
Methods Mice: 4-6 weeks old female C57BL/6 mice obtained from Jackson labs were used for cell isolation. Female Foxp3-IRES-GFP reporter mice (1), backcrossed to B6/C57 background for 10 generations, were used for the isolation of naïve CD4 and naïve CD8 cells for the RNAseq experiments. The mice were housed in pathogen-free animal facility in the La Jolla Institute for Allergy and Immunology and were used according to protocols approved by the Institutional Animal Care and use Committee. Preparation of cells: Subsets of thymocytes were isolated by cell sorting as previously described (2), after cell surface staining using CD4 (GK1.5), CD8 (53-6.7), CD3ε (145- 2C11), CD24 (M1/69) (all from Biolegend). DP cells: CD4+CD8 int/hi; CD4 SP cells: CD4CD3 hi, CD24 int/lo; CD8 SP cells: CD8 int/hi CD4 CD3 hi, CD24 int/lo (Fig S2). Peripheral subsets were isolated after pooling spleen and lymph nodes. T cells were enriched by negative isolation using Dynabeads (Dynabeads untouched mouse T cells, 11413D, Invitrogen). After surface staining for CD4 (GK1.5), CD8 (53-6.7), CD62L (MEL-14), CD25 (PC61) and CD44 (IM7), naïve CD4+CD62L hiCD25-CD44lo and naïve CD8+CD62L hiCD25-CD44lo were obtained by sorting (BD FACS Aria). Additionally, for the RNAseq experiments, CD4 and CD8 naïve cells were isolated by sorting T cells from the Foxp3- IRES-GFP mice: CD4+CD62LhiCD25–CD44lo GFP(FOXP3)– and CD8+CD62LhiCD25– CD44lo GFP(FOXP3)– (antibodies were from Biolegend). In some cases, naïve CD4 cells were cultured in vitro under Th1 or Th2 polarizing conditions (3, 4). -

Proteomic Analysis Uncovers Measles Virus Protein C Interaction with P65
bioRxiv preprint doi: https://doi.org/10.1101/2020.05.08.084418; this version posted May 9, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Proteomic Analysis Uncovers Measles Virus Protein C Interaction with p65/iASPP/p53 Protein Complex Alice Meignié1,2*, Chantal Combredet1*, Marc Santolini 3,4, István A. Kovács4,5,6, Thibaut Douché7, Quentin Giai Gianetto 7,8, Hyeju Eun9, Mariette Matondo7, Yves Jacob10, Regis Grailhe9, Frédéric Tangy1**, and Anastassia V. Komarova1, 10** 1 Viral Genomics and Vaccination Unit, Department of Virology, Institut Pasteur, CNRS UMR-3569, 75015 Paris, France 2 Université Paris Diderot, Sorbonne Paris Cité, Paris, France 3 Center for Research and Interdisciplinarity (CRI), Université de Paris, INSERM U1284 4 Network Science Institute and Department of Physics, Northeastern University, Boston, MA 02115, USA 5 Department of Physics and Astronomy, Northwestern University, Evanston, IL 60208-3109, USA 6 Department of Network and Data Science, Central European University, Budapest, H-1051, Hungary 7 Proteomics platform, Mass Spectrometry for Biology Unit (MSBio), Institut Pasteur, CNRS USR 2000, Paris, France. 8 Bioinformatics and Biostatistics Hub, Computational Biology Department, Institut Pasteur, CNRS USR3756, Paris, France 9 Technology Development Platform, Institut Pasteur Korea, Seongnam-si, Republic of Korea 10 Laboratory of Molecular Genetics of RNA Viruses, Institut Pasteur, CNRS UMR-3569, -

Host Cell Factors Necessary for Influenza a Infection: Meta-Analysis of Genome Wide Studies
Host Cell Factors Necessary for Influenza A Infection: Meta-Analysis of Genome Wide Studies Juliana S. Capitanio and Richard W. Wozniak Department of Cell Biology, Faculty of Medicine and Dentistry, University of Alberta Abstract: The Influenza A virus belongs to the Orthomyxoviridae family. Influenza virus infection occurs yearly in all countries of the world. It usually kills between 250,000 and 500,000 people and causes severe illness in millions more. Over the last century alone we have seen 3 global influenza pandemics. The great human and financial cost of this disease has made it the second most studied virus today, behind HIV. Recently, several genome-wide RNA interference studies have focused on identifying host molecules that participate in Influen- za infection. We used nine of these studies for this meta-analysis. Even though the overlap among genes identified in multiple screens was small, network analysis indicates that similar protein complexes and biological functions of the host were present. As a result, several host gene complexes important for the Influenza virus life cycle were identified. The biological function and the relevance of each identified protein complex in the Influenza virus life cycle is further detailed in this paper. Background and PA bound to the viral genome via nucleoprotein (NP). The viral core is enveloped by a lipid membrane derived from Influenza virus the host cell. The viral protein M1 underlies the membrane and anchors NEP/NS2. Hemagglutinin (HA), neuraminidase Viruses are the simplest life form on earth. They parasite host (NA), and M2 proteins are inserted into the envelope, facing organisms and subvert the host cellular machinery for differ- the viral exterior. -

High NRBP1 Expression Promotes Proliferation and Correlates With
Journal of Cancer 2019, Vol. 10 4270 Ivyspring International Publisher Journal of Cancer 2019; 10(18): 4270-4277. doi: 10.7150/jca.32656 Research Paper High NRBP1 expression promotes proliferation and correlates with poor prognosis in bladder cancer Qi Wu1,2*, Xiaoqing Zhou2*, Peng Li2, Wei Wang3, Jun Wang4, Mingyue Tan4, Le Tao1, Jianxin Qiu1 1. Department of Urology, Shanghai General Hospital of Nanjing Medical University, Shanghai 200080, China 2. Department of Urology, The Sixth Affiliated Hospital of Wenzhou Medical University (The People's Hospital of Lishui), Zhejiang 323000, P. R. China 3. Department of Urology, The Fourth Affiliated Hospital of Nantong University (Yancheng First People’s Hospital), Jiangsu 224000, P. R. China 4. Department of Urology, Shanghai General Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai 200080, P. R. China *These authors contributed equally to this work. Corresponding authors: Jianxin Qiu, Department of Urology, Shanghai General Hospital of Nanjing Medical University, No. 100 Haining Road, Hongkou District, Shanghai 200080, P. R. China. Tel: +86-13310038203; E-mail address: [email protected]; Le Tao, Department of Urology, Shanghai General Hospital of Nanjing Medical University, No. 100 Haining Road, Hongkou District, Shanghai 200080, P. R. China. Tel: +86-18817822120; Email: [email protected] © The author(s). This is an open access article distributed under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0/). See http://ivyspring.com/terms for full terms and conditions. Received: 2018.12.29; Accepted: 2019.05.12; Published: 2019.07.10 Abstract Nuclear receptor binding protein 1 (NRBP1) is an evolutionarily highly conserved adaptor protein with multiple domains. -

GILZ-Dependent Modulation of Mtorc1 Regulates Spermatogonial Maintenance Hue M
© 2018. Published by The Company of Biologists Ltd | Development (2018) 145, dev165324. doi:10.1242/dev.165324 STEM CELLS AND REGENERATION RESEARCH ARTICLE GILZ-dependent modulation of mTORC1 regulates spermatogonial maintenance Hue M. La1,2,*, Ai-Leen Chan1,2,*, Julien M. D. Legrand1,2, Fernando J. Rossello1,2, Christina G. Gangemi1,2, Antonella Papa3, Qiang Cheng4, Eric F. Morand4 and Robin M. Hobbs1,2,‡ ABSTRACT prospermatogonia) upon migration to the basement membrane of Male fertility is dependent on spermatogonial stem cells (SSCs) that the seminiferous cords. The undifferentiated population contains self-renew and produce differentiating germ cells. Growth factors isolated spermatogonia (A-single or As) plus chains of cells produced within the testis are essential for SSC maintenance interconnected by cytoplasmic bridges. Two-cell chains are but intrinsic factors that dictate the SSC response to these stimuli known as A-paired (Apr) whereas chains of four or more cells are poorly characterised. Here, we have studied the role of GILZ, are known as A-aligned (Aal). Lineage-tracing studies demonstrate α a TSC22D family protein and spermatogenesis regulator, in that SSCs are marked by GFR 1 and typically As and Apr,whereas spermatogonial function and signalling. Although broadly expressed most undifferentiated cells, particularly Aal, are committed in the germline, GILZ was prominent in undifferentiated spermatogonia progenitors and marked by NGN3 (Hara et al., 2014; Nakagawa + and Gilz deletion in adults resulted in exhaustion of the GFRα1+ et al., 2010). NGN3 Aal may revert to SSCs through chain SSC-containing population and germline degeneration. GILZ loss fragmentation, particularly upon tissue damage (Nakagawa et al., γ was associated with mTORC1 activation, suggesting enhanced 2010). -
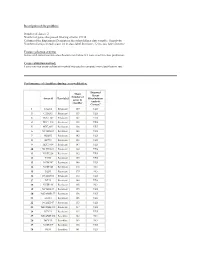
2 Number of Genes That Passed Filtering Criteria
Description of the problem: Number of classes: 2 Number of genes that passed filtering criteria: 25338 Column of the Experiment Descriptors sheet that defines class variable: Sensitivity Number of arrays in each class: 24 in class label Resistant , 32 in class label Sensitive Feature selection criteria: Genes with univariate mis-classfication rate below 0.2 were used for class prediction. Cross-validation method: Leave-one-out cross-validation method was used to compute mis-classification rate. Performance of classifiers during cross-validation. Diagonal Mean Linear Number of Array id Class label Discriminant genes in Analysis classifier Correct? 1 CALU1 Resistant 129 YES 2 CAMA1 Resistant 155 YES 3 HCC1143 Resistant 145 YES 4 HCC1419 Resistant 148 YES 5 HCC2935 Resistant 136 YES 6 NCIH2405 Resistant 148 YES 7 SKBR3 Resistant 142 YES 8 ZR751 Resistant 133 YES 9 HCC1954 Resistant 147 YES 10 NCIH1651 Resistant 139 YES 11 NCIH226 Resistant 142 YES 12 T47D Resistant 139 YES 13 NCIH747 Resistant 146 YES 14 NCIH345 Resistant 216 NO 15 DLD1 Resistant 179 NO 16 NCIH1666 Resistant 154 YES 17 BT20 Resistant 140 YES 18 NCIH196 Resistant 168 NO 19 NCIH1437 Resistant 159 YES 20 MDAMB157 Resistant 158 YES 21 LS123 Resistant 155 YES 22 NCIH2347 Resistant 133 YES 23 MDAMB361 Resistant 137 YES 24 BT474 Resistant 155 YES 25 MDAMB468 Sensitive 182 NO 26 MCF10 Sensitive 169 NO 27 NCIH209 Sensitive 128 YES 28 RKO Sensitive 141 YES 29 HCC70 Sensitive 182 NO 30 LS513 Sensitive 136 YES 31 NCIH1963 Sensitive 122 YES 32 HCC1187 Sensitive 145 YES 33 T84 Sensitive -

1 Trisomy 21 Consistently Activates the Interferon Response Kelly D
1 2 Trisomy 21 consistently activates the interferon response 3 4 Kelly D. Sullivan1,2,3,4*, Hannah C. Lewis1,2, Amanda A. Hill1,2, Ahwan Pandey1,2,3,4, Leisa P. 5 Jackson1,3,4, Joseph M. Cabral1,3,4, Keith P. Smith1, L. Alexander Liggett1,5, Eliana B. Gomez1,3,4, 6 Matthew D. Galbraith1,2,3,4, James DeGregori1,5,6, and Joaquín M. Espinosa1,2,3,4* 7 8 1Linda Crnic Institute for Down Syndrome, University of Colorado School of Medicine, Aurora, 9 Colorado 80045, USA 10 2Department of Pharmacology, University of Colorado School of Medicine, Aurora, Colorado 11 80045, USA 12 3Department of Molecular, Cellular and Developmental Biology, University of Colorado Boulder, 13 Boulder, Colorado 80203, USA 14 4Howard Hughes Medical Institute, Chevy Chase, Maryland 20815, USA 15 5Department of Biochemistry and Molecular Genetics, University of Colorado School of 16 Medicine, Aurora, Colorado 80045, USA 17 6Department of Pediatrics, Integrated Department of Immunology, Section of Hematology, 18 Department of Medicine, University of Colorado School of Medicine, Aurora, CO 80045. 19 20 21 *Correspondence to: [email protected] / [email protected] 22 23 1 24 ABSTRACT 25 Although it is clear that trisomy 21 causes Down syndrome, the molecular events acting 26 downstream of the trisomy remain ill defined. Using complementary genomics analyses, we 27 identified the interferon pathway as the major signaling cascade consistently activated by 28 trisomy 21 in human cells. Transcriptome analysis revealed that trisomy 21 activates the 29 interferon transcriptional response in fibroblast and lymphoblastoid cell lines, as well as 30 circulating monocytes and T cells. -

Chromatin Conformation Links Distal Target Genes to CKD Loci
BASIC RESEARCH www.jasn.org Chromatin Conformation Links Distal Target Genes to CKD Loci Maarten M. Brandt,1 Claartje A. Meddens,2,3 Laura Louzao-Martinez,4 Noortje A.M. van den Dungen,5,6 Nico R. Lansu,2,3,6 Edward E.S. Nieuwenhuis,2 Dirk J. Duncker,1 Marianne C. Verhaar,4 Jaap A. Joles,4 Michal Mokry,2,3,6 and Caroline Cheng1,4 1Experimental Cardiology, Department of Cardiology, Thoraxcenter Erasmus University Medical Center, Rotterdam, The Netherlands; and 2Department of Pediatrics, Wilhelmina Children’s Hospital, 3Regenerative Medicine Center Utrecht, Department of Pediatrics, 4Department of Nephrology and Hypertension, Division of Internal Medicine and Dermatology, 5Department of Cardiology, Division Heart and Lungs, and 6Epigenomics Facility, Department of Cardiology, University Medical Center Utrecht, Utrecht, The Netherlands ABSTRACT Genome-wide association studies (GWASs) have identified many genetic risk factors for CKD. However, linking common variants to genes that are causal for CKD etiology remains challenging. By adapting self-transcribing active regulatory region sequencing, we evaluated the effect of genetic variation on DNA regulatory elements (DREs). Variants in linkage with the CKD-associated single-nucleotide polymorphism rs11959928 were shown to affect DRE function, illustrating that genes regulated by DREs colocalizing with CKD-associated variation can be dysregulated and therefore, considered as CKD candidate genes. To identify target genes of these DREs, we used circular chro- mosome conformation capture (4C) sequencing on glomerular endothelial cells and renal tubular epithelial cells. Our 4C analyses revealed interactions of CKD-associated susceptibility regions with the transcriptional start sites of 304 target genes. Overlap with multiple databases confirmed that many of these target genes are involved in kidney homeostasis. -

Page 1 Exploring the Understudied Human Kinome For
bioRxiv preprint doi: https://doi.org/10.1101/2020.04.02.022277; this version posted June 30, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. Exploring the understudied human kinome for research and therapeutic opportunities Nienke Moret1,2,*, Changchang Liu1,2,*, Benjamin M. Gyori2, John A. Bachman,2, Albert Steppi2, Rahil Taujale3, Liang-Chin Huang3, Clemens Hug2, Matt Berginski1,4,5, Shawn Gomez1,4,5, Natarajan Kannan,1,3 and Peter K. Sorger1,2,† *These authors contributed equally † Corresponding author 1The NIH Understudied Kinome Consortium 2Laboratory of Systems Pharmacology, Department of Systems Biology, Harvard Program in Therapeutic Science, Harvard Medical School, Boston, Massachusetts 02115, USA 3 Institute of Bioinformatics, University of Georgia, Athens, GA, 30602 USA 4 Department of Pharmacology, The University of North Carolina at Chapel Hill, Chapel Hill, NC 27599, USA 5 Joint Department of Biomedical Engineering at the University of North Carolina at Chapel Hill and North Carolina State University, Chapel Hill, NC 27599, USA Key Words: kinase, human kinome, kinase inhibitors, drug discovery, cancer, cheminformatics, † Peter Sorger Warren Alpert 432 200 Longwood Avenue Harvard Medical School, Boston MA 02115 [email protected] cc: [email protected] 617-432-6901 ORCID Numbers Peter K. Sorger 0000-0002-3364-1838 Nienke Moret 0000-0001-6038-6863 Changchang Liu 0000-0003-4594-4577 Ben Gyori 0000-0001-9439-5346 John Bachman 0000-0001-6095-2466 Albert Steppi 0000-0001-5871-6245 Page 1 bioRxiv preprint doi: https://doi.org/10.1101/2020.04.02.022277; this version posted June 30, 2020. -

Predict AID Targeting in Non-Ig Genes Multiple Transcription Factor
Downloaded from http://www.jimmunol.org/ by guest on September 26, 2021 is online at: average * The Journal of Immunology published online 20 March 2013 from submission to initial decision 4 weeks from acceptance to publication Multiple Transcription Factor Binding Sites Predict AID Targeting in Non-Ig Genes Jamie L. Duke, Man Liu, Gur Yaari, Ashraf M. Khalil, Mary M. Tomayko, Mark J. Shlomchik, David G. Schatz and Steven H. Kleinstein J Immunol http://www.jimmunol.org/content/early/2013/03/20/jimmun ol.1202547 Submit online. Every submission reviewed by practicing scientists ? is published twice each month by http://jimmunol.org/subscription Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts http://www.jimmunol.org/content/suppl/2013/03/20/jimmunol.120254 7.DC1 Information about subscribing to The JI No Triage! Fast Publication! Rapid Reviews! 30 days* Why • • • Material Permissions Email Alerts Subscription Supplementary The Journal of Immunology The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2013 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. This information is current as of September 26, 2021. Published March 20, 2013, doi:10.4049/jimmunol.1202547 The Journal of Immunology Multiple Transcription Factor Binding Sites Predict AID Targeting in Non-Ig Genes Jamie L. Duke,* Man Liu,†,1 Gur Yaari,‡ Ashraf M. Khalil,x Mary M. Tomayko,{ Mark J. Shlomchik,†,x David G. -

Nuclear Receptor Binding Protein 1 Regulates Intestinal Progenitor Cell Homeostasis and Tumour Formation
Nuclear Receptor Binding Protein 1 Regulates Intestinal Progenitor Cell Homeostasis and Tumour Formation The Harvard community has made this article openly available. Please share how this access benefits you. Your story matters Citation Wilson, Catherine H., Catriona Crombie, Louise van der Weyden, George Poulogiannis, Alistair G. Rust, Mercedes Pardo, Tannia Gracia, et al. 2012. Nuclear receptor binding protein 1 regulates intestinal progenitor cell homeostasis and tumour formation. The EMBO Journal 31(11): 2486-2497. Published Version doi:10.1038/emboj.2012.91 Citable link http://nrs.harvard.edu/urn-3:HUL.InstRepos:10417530 Terms of Use This article was downloaded from Harvard University’s DASH repository, and is made available under the terms and conditions applicable to Other Posted Material, as set forth at http:// nrs.harvard.edu/urn-3:HUL.InstRepos:dash.current.terms-of- use#LAA The EMBO Journal (2012) 31, 2486–2497 | & 2012 European Molecular Biology Organization | Some Rights Reserved 0261-4189/12 www.embojournal.org TTHEH E EEMBOMBO JJOURNALOURN AL Nuclear receptor binding protein 1 regulates EMBO intestinal progenitor cell homeostasis and tumour open formation Catherine H Wilson1,7, Catriona Crombie1, Introduction Louise van der Weyden1, 2 1 Perturbation of RAS, WNT or NOTCH signalling in the George Poulogiannis , Alistair G Rust , C. elegans vulva results in defects in vulval development, Mercedes Pardo1, Tannia Gracia1,8,LuYu1, 1 3 and many genetic screens have exploited this fact to identify Jyoti Choudhary , Gino B Poulin , important components of these pathways. In humans, the 1 4 Rebecca E McIntyre , Douglas J Winton , RAS, WNT and NOTCH pathways play an important role in 4 5,9 H Nikki March , Mark J Arends , orchestrating embryonic development and may be dysregu- 1,6,9 1,9, Andrew G Fraser and David J Adams * lated in tumourigenesis.