Formal Synthesis of Palmerolide a Using Fragmentation Methodology Marilda Pereira Lisboa
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Supporting Information a Convenient Chromatography-Free Method For
Electronic Supplementary Material (ESI) for Organic & Biomolecular Chemistry This journal is © The Royal Society of Chemistry 2012 Supporting Information A Convenient Chromatography-Free Method for the Purification of Alkenes Produced in the Wittig Reaction Peter A. Byrne, Kamalraj V. Rajendran, Jimmy Muldoon and Declan G. Gilheany Centre for Synthesis and Chemical Biology, School of Chemistry and Chemical Biology, University College Dublin, Belfield, Dublin 4, Ireland 1. General Experimental 2 2. Synthesis of phosphonium salts 4 3. Procedures for Wittig reactions & phosphine oxide removal 11 4. Characterisation of purified alkenes 15 5. Reduction of phosphine chalcogenides 34 6. Synthesis & isolation of neomenthyl chloride and reduction of phosphine oxide-by-product 37 7. NMR spectra of phosphonium salts 42 8. NMR spectra of purified alkenes and regenerated phosphines from Wittig reactions 56 9. NMR spectra of phosphines produced by reduction of phosphine chalcogenides 85 10. NMR spectra of purified products from Appel-type reactions 90 11. References 91 1 Electronic Supplementary Material (ESI) for Organic & Biomolecular Chemistry This journal is © The Royal Society of Chemistry 2012 1. General Experimental All chemicals were supplied by Aldrich, with the exception of Zeoprep silica, 2- methylbenzaldehyde ( o-tolualdehyde, Fluka), ( tert -butoxycarbonylmethyl)- -1 triphenylphosphonium bromide (Fluka), 1 mol L LiAlH 4 in THF (Acros Organics) and Merck standardised alumina 90. All chemicals were used without further purification except diethyl ether, toluene, and THF, which were processed through an Innovative Technology Inc. Pure Solv-400-3-MD solvent purification (Grubbs still) system and stored in Strauss flasks under a nitrogen atmosphere, and ethyl acetate and dichloromethane, which were degassed by passing a stream of dry nitrogen gas (oxygen- free) through the solvent for one hour for the purposes of work-ups in phosphine syntheses. -

Brittain-DR-1965-Phd-Thesis.Pdf
POLYMETHYLENE PYRIDINES , A thesis submitted by David Robert Brittain in partial fulfilment of the requirements for the degree of DOCTOR OP PHILOSOPHY in the University of London Organic Chemistry Department, dune, 1965. Imperial College, LONDON, S.W.7. ABSTRACT This thesis describes a series of attempts to syn- thesise 2,5- and 1,4-polymethylene bridged pyridines. Nuclear magnetic resonance theory predicts that protons, which are held directly over an aromatic ring, will be abnormally shielded compared with protons in aliphatic straight-chain hydrocarbons. This prediction has been verified for the central methylene protons of paracyclo- phanes. The degree of shielding, expressed in terms of the distance from the aromatic ring, is a measure of the induced ring current and hence the aromaticity of the benzene ring. Similar measurements upon 2,5- or 1,4— polymethylene bridged pyridines would make it possible to determine the degree of aromaticity of the pyridine ring relative to benzene. A review of the subject of aromaticity is presented in which special reference has been made to its inter- pretation by nuclear magnetic resonance. The synthetic work has not been brougL.t to a truly satisfactory conclusion. However, the synthetic routes to 2,5-dialkylpyridines have been thoroughly investigated and a wide variety of such compounds prepared. The functional groups at the ends of the alkyl chains have been varied in an effort to produce a derivative which would cyclise to give a 2,5-bridged pyridine. The attempted intramolecular oxidative coupling of 2,5-dihex- 51 -ynylpyridine received much attention. In the attempts to obtain a 1,4-bridged pyridine, two tricyclic compounds, each containing two quaternised pyridine rings linked by polymethylene chains, were obtained. -

Synthesis of the Methyl Ester Of
Master’s Thesis 2019 60 ECTS Faculty of Chemistry, Biotechnology and Food Science Synthesis of the Methyl Ester of MaR2n-3 DPA Jeanne Sønderskov Rasmussen Master’s degree in Chemistry and Biotechnology II Acknowledgements The master’s degree was performed as collaboration between the Faculty of Chemistry, Biotechnology and Food Science at the Norwegian University of Life Sciences and the Department of Pharmacy, University of Oslo. The practical work as part of this thesis was conducted in the LIPCHEM group at the Section of Pharmaceutical Chemistry. First, I would like to thank my two supervisors, Professor Trond Vidar Hansen and Professor Yngve Stenstrøm for excellent guidance, encouragement and especially, for sharing long-time experience. Also, thanks to Dr. Jørn Tungen and Associate professor Anders Vik for solid guidance and teaching, especially during the time in the laboratory. I would also like to thank the rest of the LIPCHEM group. It has been a great experience to be a part of a research group and witness the impressive work that is carried out. Also, I am very grateful for sharing this challenging period with the four master students in pharmaceutical chemistry, Amalie Føreid Reinertsen, Marie Hermansen Mørk, Aina Kristin Pham and Margrethe Kristiansen. It has been nice to get to know you and follow your progress as well. Finally, an enormous thank to my lovely boyfriend, family and friends. Thanks for fantastic support and cheering through my six years of education. Blindern, May 2019 Jeanne Sønderskov Rasmussen III Abstract This master thesis presents the first synthesis of the methyl ester of the specialized pro-resolving mediator named MaR2n-3 DPA. -

Syntheses and Eliminations of Cyclopentyl Derivatives David John Rausch Iowa State University
Iowa State University Capstones, Theses and Retrospective Theses and Dissertations Dissertations 1966 Syntheses and eliminations of cyclopentyl derivatives David John Rausch Iowa State University Follow this and additional works at: https://lib.dr.iastate.edu/rtd Part of the Organic Chemistry Commons Recommended Citation Rausch, David John, "Syntheses and eliminations of cyclopentyl derivatives " (1966). Retrospective Theses and Dissertations. 2875. https://lib.dr.iastate.edu/rtd/2875 This Dissertation is brought to you for free and open access by the Iowa State University Capstones, Theses and Dissertations at Iowa State University Digital Repository. It has been accepted for inclusion in Retrospective Theses and Dissertations by an authorized administrator of Iowa State University Digital Repository. For more information, please contact [email protected]. This dissertation has been microfilmed exactly as received 66—6996 RAUSCH, David John, 1940- SYNTHESES AND ELIMINATIONS OF CYCLOPENTYL DERIVATIVES. Iowa State University of Science and Technology Ph.D., 1966 Chemistry, organic University Microfilms, Inc., Ann Arbor, Michigan SYNTHESES AND ELIMINATIONS OF CYCLOPENTYL DERIVATIVES by David John Rausch A Dissertation Submitted to the Graduate Faculty in Partial Fulfillment of The Requirements for the Degree of DOCTOR OF PHILOSOPHY Major Subject: Organic Chemistry Approved : Signature was redacted for privacy. Signature was redacted for privacy. Head of Major Department Signature was redacted for privacy. Iowa State University Of Science and Technology Ames, Iowa 1966 ii TABLE OF CONTENTS VITA INTRODUCTION HISTORICAL Conformation of Cyclopentanes Elimination Reactions RESULTS AND DISCUSSION Synthetic Elimination Reactions EXPERIMENTAL Preparation and Purification of Materials Procedures and Data for Beta Elimination Reactions SUMMARY LITERATURE CITED ACKNOWLEDGEMENTS iii VITA The author was born in Aurora, Illinois, on October 24, 1940, to Mr. -

1St Year Students Lecture No. 5 Ahmed Mohamad Jawad
1st year students Lecture No. 5 Ahmed Mohamad Jawad Create PDF files without this message by purchasing novaPDF printer (http://www.novapdf.com) Out line 1- Dienes: Introduction of diene Nomenclature of diene Preparation of diene Reaction of diene 2- Alkyne Introduction of Alkyne Nomenclature of Alkyne Physical properties of alkyne Preparation of Alkyne Reaction of Alkyne Create PDF files without this message by purchasing novaPDF printer (http://www.novapdf.com) Introduction of diene Dienes are simply alkenes that contain two carbon-carbon double bonds. Dienes are divided into two major important classes according to the arrangement of the double bonds 1-Conjugated : Double bonds that alternate with single bonds are said to be conjugated. Create PDF files without this message by purchasing novaPDF printer (http://www.novapdf.com) Introduction of diene 2-Isolated double bonds that are separated by more than one single bond are said to be isolated Less stable than conjugated 3-Cumulated : contains cumulated double bonds Least one stable . Create PDF files without this message by purchasing novaPDF printer (http://www.novapdf.com) Nomenclature of diene Dienes are named by the IUPAC system in the same way as alkenes , except that the ending diene is used, with two numbers to indicate the position of the two double bonds. H2C C CH2 1,2-propadiene 1,3-butadiene 1,4-pentadiene This system is easily extended to compounds containing any number of double bonds. Create PDF files without this message by purchasing novaPDF printer (http://www.novapdf.com) Preparation of diene 1- by acid catalyzed double dehydration 2- By dehydrogenation of dihalides: Create PDF files without this message by purchasing novaPDF printer (http://www.novapdf.com) Reactions of Dienes In general terms, dienes undergo electrophilic addition reactions in a similar approach of alkenes Conjugated dienes undergo addition but the proximity of the conjugated C=C influences the reactions. -

Photoremovable Protecting Groups Used for the Caging of Biomolecules
1 1 Photoremovable Protecting Groups Used for the Caging of Biomolecules 1.1 2-Nitrobenzyl and 7-Nitroindoline Derivatives John E.T. Corrie 1.1.1 Introduction 1.1.1.1 Preamble and Scope of the Review This chapter covers developments with 2-nitrobenzyl (and substituted variants) and 7-nitroindoline caging groups over the decade from 1993, when the author last reviewed the topic [1]. Other reviews covered parts of the field at a similar date [2, 3], and more recent coverage is also available [4–6]. This chapter is not an exhaustive review of every instance of the subject cages, and its principal focus is on the chemistry of synthesis and photocleavage. Applications of indi- vidual compounds are only briefly discussed, usually when needed to put the work into context. The balance between the two cage types is heavily slanted to- ward the 2-nitrobenzyls, since work with 7-nitroindoline cages dates essentially from 1999 (see Section 1.1.3.2), while the 2-nitrobenzyl type has been in use for 25 years, from the introduction of caged ATP 1 (Scheme 1.1.1) by Kaplan and co-workers [7] in 1978. Scheme 1.1.1 Overall photolysis reaction of NPE-caged ATP 1. Dynamic Studies in Biology. Edited by M. Goeldner, R. Givens Copyright © 2005 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim ISBN: 3-527-30783-4 2 1 Photoremovable Protecting Groups Used for the Caging of Biomolecules 1.1.1.2 Historical Perspective The pioneering work of Kaplan et al. [7], although preceded by other examples of 2-nitrobenzyl photolysis in synthetic organic chemistry, was the first to apply this to a biological problem, the erythrocytic Na:K ion pump. -

Diels-Alder Reaction Between Naphthalene and N-Phenylmaleimide Under Ambient and High Pressure Conditions
Issue in Honor of Prof. Alexander I. Konovalov ARKIVOC 2004 (xii) 70-79 Diels-Alder reaction between naphthalene and N-phenylmaleimide under ambient and high pressure conditions Gulnara G. Iskhakova*, Vladimir D. Kiselev, Elena A. Kashaeva, Lubov’ N. Potapova, Evgeny A. Berdnikov, Dmitry B. Krivolapov, and Igor A. Litvinov A. M. Butlerov Chemical Institute, Kazan State University, Kremlevskaya str.18, 420008 Kazan, Russian Federation E-mail: [email protected] Dedicated to the 70th anniversary of Professor Alexander I. Konovalov (received 02 Nov 04; accepted 08 Jan 05; published on the web 05 Feb 05) Abstract The rate and equilibrium constants for the Diels-Alder reactions between benzene and naphthalene as dienes and tetracyanoethylene, maleic anhydride and N-phenylmaleimide as dienophiles at 25 °С were estimated from empirical rule. The highest yield of the adduct was predicted for the reaction of naphthalene with N-phenylmaleimide. The time of adduct formation in 50% yield exceeds 30 years. The use of gallium chloride as a catalyst affords the exo-adduct for seven days at room temperature. The rate ((2±0.5)•10-6 L mol-1 s-1) and equilibrium constants (5±2 L mol-1) of this reaction were determined. Under high pressure conditions (8 kbar) reaction occurs with formation of both stereo isomers at 100 °С during 80 hours. Keywords: Naphthalene, Diels-Alder reaction, catalysis, high pressure Introduction 1 Many dienes, including substituted benzenes and naphthalenes, are known to form molecular complexes of the π,π-type due to the interaction of the highest occupied π-orbital of a diene- donor with the lowest unoccupied π-orbital of a dienophile-acceptor. -

19.13 the Wittig Alkene Synthesis 933
19_BRCLoudon_pgs5-0.qxd 12/9/08 11:41 AM Page 933 19.13 THE WITTIG ALKENE SYNTHESIS 933 PROBLEMS 19.32 Draw the structures of all aldehydes or ketones that could in principle give the following product after application of either the Wolff–Kishner or Clemmensen reduction. H3C CH2CH(CH3)2 L L 19.33 Outline a synthesis of 1,4-dimethoxy-2-propylbenzene from hydroquinone (p-hydroxyphenol) and any other reagents. 19.13 THE WITTIG ALKENE SYNTHESIS Our tour through aldehyde and ketone chemistry started with simple additions; then addition followed by substitution (acetal formation); then additions followed by elimination (imine and enamine formation). Another addition–elimination reaction, called the Wittig alkene synthe- sis, is an important method for preparing alkenes from aldehydes and ketones. An example of the Wittig alkene synthesis is the preparation of methylenecyclohexane from cyclohexanone. 1 1 A | A | O CH_ 2 PPh3 CH2 Ph3P O _ (19.70) 1 + 3 anL ylid + L 1 3 triphenylphosphine cyclohexanone methylenecyclohexane oxide The Wittig synthesis is especially important because it gives alkenes in which the position of the double bond is unambiguous; in other words, the Wittig synthesis is completely regios- elective.Itcanbeusedforthepreparationofalkenesthatwouldbedifficulttoprepareby other reactions. For example, methylenecyclohexane, which is readily prepared by the Wittig synthesis (Eq. 19.70), cannot be prepared by dehydration of 1-methylcyclohexanol; 1- methylcyclohexene is obtained instead, because alcohol dehydration gives the alkene iso- mer(s) in which the double bond has the greatest number of alkyl substituents (Sec. 10.1). OH " H2SO4 " CH3 H2O CH3 L + 1-methylcyclohexene H2SO4 (19.71) A CH2 (little or none formed) methylenecyclohexane The nucleophile in the Wittig alkene synthesis is a type of ylid. -
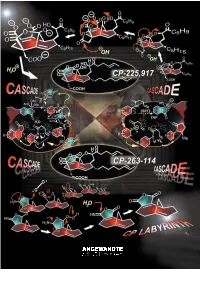
A Paradigm of How Endeavors in Total Synthesis Lead to Discoveries and Inventions in Organic Synthesis
REVIEWS The CP Molecule Labyrinth: A Paradigm of How Endeavors in Total Synthesis Lead to Discoveries and Inventions in Organic Synthesis K. C. Nicolaou* and Phil S. Baran Dedicated to Mrs. Niki Goulandris for her outstanding contributions to humanity and Planet Earth on the occasion of the opening of the GAIA Center for Environmental Research and Education at the Goulandris Natural History Museum in Athens, Greece. Imagine an artist carving a sculpture Herculean nature of the task and the ed Minotaur, which he accomplished from a marble slab and finding gold rewards that accompany it, one must through brilliance, skill, and bravery nuggets in the process. This thought is sense the details of the enterprise having traversed the famous labyrinth not a far-fetched description of the behind the scenes. A more vivid de- with the help of Ariadne. This story work of a synthetic chemist pursuing scription of total synthesis as a struggle from Greek mythology comes alive in the total synthesis of a natural product. against a tough opponent is perhaps modern synthetic expeditions toward At the end of the day, he or she will be appropriate to dramatize these ele- natural products as exemplified by the judged by the artistry of the final work ments of the experience. In this article total synthesis of the CP molecules and the weight of the gold discovered we describe one such endeavor of total which serve as a paradigm for modern in the process. However, as colorful as synthesis which, in addition to reaching total synthesis endeavors, where the this description of total synthesis may the target molecule, resulted in a objectives are discovery and invention be, it does not entirely capture the wealth of new synthetic strategies and in the broader sense of organic syn- essence of the endeavor, for there is technologies for chemical synthesis. -

Industrial Chemistry Process Scheme for Sections 3 - 3.3.2 Butenes Natural Gas Lwet I Oil
Olefins - Part 1 Industrial Chemistry Process Scheme for Sections 3 - 3.3.2 Butenes Natural Gas lwet I Oil Gas Gas Naphtha Oil Vacuum Distillation iiHydrotreating Naphtha Catalytic and Steam Thermal Crack- Process &--Crac kin ------l Where they come from Ethylenenm Propene Acetylene Ethylene Fuel Gas Ethylene Propene CL- C5- Pyrolysis Fraction Fraction Gasoline , + I 112.2.1 CL-Riffinate C5- Raffinate Ethylene 2-Butene manufacture Ratfinate I1 isobutene or n- Butenes n- and lsopentenes n-Pentenes lsobutene Oligomers n- and lsobutane n- and lsopentanes n- and lsopentanes 68 3. Olejins most valuable C4 olefin source: As shown in Table 3-3, a significantly higher fraction of steam cracking of naphtha butenes is obtained from steam cracking of naphtha than from Which C4 are obtainedcatalytic by cracking steam of gas oil. Therefore cracking? naphtha steam cracking is the more interesting technology for production of unsatu- rated C4 compounds. decrease of total C4 and C4 olefins, but As the cracking- severity increases, both the total yield of the increase Of butadiene under high severity C4 fraction and the proportion of butenes decrease, while the cracking conditions (consequence of butanedesired higher C2H4isobutane yield) proportion of butadiene increases due to its higher stability: Table 3-3. Composition of Cq fractions from steam cracking of naphtha and catalytic cracking of gas oil (in wt%). but-1-ene (E)-but-2-ene Steam cracking Catalytic cracking 1-butene Cracked products Low High (FCC) zeolite trans-2-butene severity seventy catalyst -

Appendix I: Named Reactions Single-Bond Forming Reactions Co
Appendix I: Named Reactions 235 / 335 432 / 533 synthesis / / synthesis Covered in Covered Featured in problem set problem Single-bond forming reactions Grignard reaction various Radical couplings hirstutene Conjugate addition / Michael reaction strychnine Stork enamine additions Aldol-type reactions (incl. Mukaiyama aldol) various (aldol / Claisen / Knoevenagel / Mannich / Henry etc.) Asymmetric aldol reactions: Evans / Carreira etc. saframycin A Organocatalytic asymmetric aldol saframycin A Pseudoephedrine glycinamide alkylation saframycin A Prins reaction Prins-pinacol reaction problem set # 2 Morita-Baylis-Hillman reaction McMurry condensation Gabriel synthesis problem set #3 Double-bond forming reactions Wittig reaction prostaglandin Horner-Wadsworth-Emmons reaction prostaglandin Still-Gennari olefination general discussion Julia olefination and heteroaryl variants within the Corey-Winter olefination prostaglandin Peterson olefination synthesis Barton extrusion reaction Tebbe olefination / other methylene-forming reactions tetrodotoxin hirstutene / Selenoxide elimination tetrodotoxin Burgess dehydration problem set # 3 Electrocyclic reactions and related transformations Diels-Alder reaction problem set # 1 Asymmetric Diels-Alder reaction prostaglandin Ene reaction problem set # 3 1,3-dipolar cycloadditions various [2,3] sigmatropic rearrangement various Cope rearrangement periplanone Claisen rearrangement hirstutene Oxidations – Also See Handout # 1 Swern-type oxidations (Swern / Moffatt / Parikh-Doering etc. N1999A2 Jones oxidation -

Studies in Directed Catalytic Asymmetric Hydroboration of 1,2-Disubstituted Unsaturated Amide
University of Nebraska - Lincoln DigitalCommons@University of Nebraska - Lincoln Student Research Projects, Dissertations, and Chemistry, Department of Theses - Chemistry Department Summer 7-2017 STUDIES IN DIRECTED CATALYTIC ASYMMETRIC HYDROBORATION OF 1,2-DISUBSTITUTED UNSATURATED AMIDE Shuyang Zhang University of Nebraska - Lincoln, [email protected] Follow this and additional works at: http://digitalcommons.unl.edu/chemistrydiss Part of the Organic Chemistry Commons Zhang, Shuyang, "STUDIES IN DIRECTED CATALYTIC ASYMMETRIC HYDROBORATION OF 1,2-DISUBSTITUTED UNSATURATED AMIDE" (2017). Student Research Projects, Dissertations, and Theses - Chemistry Department. 85. http://digitalcommons.unl.edu/chemistrydiss/85 This Article is brought to you for free and open access by the Chemistry, Department of at DigitalCommons@University of Nebraska - Lincoln. It has been accepted for inclusion in Student Research Projects, Dissertations, and Theses - Chemistry Department by an authorized administrator of DigitalCommons@University of Nebraska - Lincoln. STUDIES IN DIRECTED CATALYTIC ASYMMETRIC HYDROBORATION OF 1,2- DISUBSTITUTED UNSATURATED AMIDE by Shuyang Zhang A THESIS Presented to the Faculty of The Graduate College at the University of Nebraska In Partial Fulfillment of Requirements For the Degree of Master of Science Major: Chemistry Under the Supervision of Professor James M. Takacs Lincoln, Nebraska July 2017 DIRECTED CATALYTIC ASYMMETRIC HYDROBORATION OF 1,2- DISUBSTITUTED ALKENES Shuyang Zhang, M.S. University of Nebraska 2017 Advisor: Professor James M. Takacs The Rh-catalyzed, substrate directed catalytic asymmetric hydroboration of γ,δ-unsaturated amides provides a direct route to enantioenriched acyclic secondary γ- borylated carbonyl derivatives with high regio- and enantioselectivity. The catalytic condition optimization and substrate scope study is discussed, including the effects in catalytical asymmetric hydroboration on pre-installed chiral γ,δ-unsaturated amides.