Characterization of a Y‐Specific Duplication/Insertion of the Anti
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Widespread Cis-Regulatory Convergence Between the Extinct Tasmanian Tiger and Gray Wolf
Downloaded from genome.cshlp.org on October 7, 2021 - Published by Cold Spring Harbor Laboratory Press Research Widespread cis-regulatory convergence between the extinct Tasmanian tiger and gray wolf Charles Y. Feigin,1,2 Axel H. Newton,1,3 and Andrew J. Pask1,3 1School of BioSciences, The University of Melbourne, Parkville, Victoria 3010, Australia; 2Department of Molecular Biology, Princeton University, Princeton, New Jersey 08544, USA; 3Museums Victoria, Melbourne, Victoria 3053, Australia The extinct marsupial Tasmanian tiger, or thylacine, and the eutherian gray wolf are among the most widely recognized examples of convergent evolution in mammals. Despite being distantly related, these large predators independently evolved extremely similar craniofacial morphologies, and evidence suggests that they filled similar ecological niches. Previous anal- yses revealed little evidence of adaptive convergence between their protein-coding genes. Thus, the genetic basis of their convergence is still unclear. Here, we identified candidate craniofacial cis-regulatory elements across vertebrates and com- pared their evolutionary rates in the thylacine and wolf, revealing abundant signatures of convergent positive selection. Craniofacial thylacine–wolf accelerated regions were enriched near genes involved in TGF beta (TGFB) and BMP signaling, both of which are key morphological signaling pathways with critical roles in establishing the identities and boundaries be- tween craniofacial tissues. Similarly, enhancers of genes involved in craniofacial nerve development showed convergent se- lection and involvement in these pathways. Taken together, these results suggest that adaptation in cis-regulators of TGF beta and BMP signaling may provide a mechanism to explain the coevolution of developmentally and functionally integrat- ed craniofacial structures in these species. -

Decorin Knockdown Affects the Gene Expression Profile of Adhesion, Growth and Extracellular Matrix Metabolism in C-28/I2 Chondrocytes
PLOS ONE RESEARCH ARTICLE Decorin knockdown affects the gene expression profile of adhesion, growth and extracellular matrix metabolism in C-28/I2 chondrocytes ☯ ☯ Mengying WangID , Zhengzheng Li , Meng Zhang, Hui Wang, Ying Zhang, Yiping Feng, Yinan Liu, Jinghong Chen*☯ Institute of Endemic Diseases, School of Public Health, Xi'an Jiaotong University Health Science Center, a1111111111 Xi'an, Shaanxi, PR China a1111111111 a1111111111 ☯ These authors contributed equally to this work. a1111111111 * [email protected] a1111111111 Abstract Decorin is a member of small leucine-rich proteoglycan family, which is involved in multiple OPEN ACCESS biological functions mainly as a structural and signaling molecule, and disturbances in its Citation: Wang M, Li Z, Zhang M, Wang H, Zhang own metabolism plays a crucial role in the pathogenesis of osteoarthropathy. In this study, Y, Feng Y, et al. (2020) Decorin knockdown affects we aim to further explore the biological function of decorin and their role in human chondro- the gene expression profile of adhesion, growth cyte cell line, C28/I2. Lentivirus-mediated shRNA was applied to down-regulate decorin and extracellular matrix metabolism in C-28/I2 chondrocytes. PLoS ONE 15(4): e0232321. https:// expression in C28/I2 chondrocytes. Effect of decorin knockdown on gene expression pro- doi.org/10.1371/journal.pone.0232321 files was determined by RNA sequencing followed by bioinformatics analysis. MTT, adhe- Editor: Dragana Nikitovic, University of Crete, sion assays and flow cytometry were used to investigate the effect of decorin knockdown on GREECE cell proliferation, adhesion, and apoptosis. sGAG content in the culture medium was deter- Received: January 2, 2020 mined by DMMB assay. -

Supplementary Materials
Supplementary Materials + - NUMB E2F2 PCBP2 CDKN1B MTOR AKT3 HOXA9 HNRNPA1 HNRNPA2B1 HNRNPA2B1 HNRNPK HNRNPA3 PCBP2 AICDA FLT3 SLAMF1 BIC CD34 TAL1 SPI1 GATA1 CD48 PIK3CG RUNX1 PIK3CD SLAMF1 CDKN2B CDKN2A CD34 RUNX1 E2F3 KMT2A RUNX1 T MIXL1 +++ +++ ++++ ++++ +++ 0 0 0 0 hematopoietic potential H1 H1 PB7 PB6 PB6 PB6.1 PB6.1 PB12.1 PB12.1 Figure S1. Unsupervised hierarchical clustering of hPSC-derived EBs according to the mRNA expression of hematopoietic lineage genes (microarray analysis). Hematopoietic-competent cells (H1, PB6.1, PB7) were separated from hematopoietic-deficient ones (PB6, PB12.1). In this experiment, all hPSCs were tested in duplicate, except PB7. Genes under-expressed or over-expressed in blood-deficient hPSCs are indicated in blue and red respectively (related to Table S1). 1 C) Mesoderm B) Endoderm + - KDR HAND1 GATA6 MEF2C DKK1 MSX1 GATA4 WNT3A GATA4 COL2A1 HNF1B ZFPM2 A) Ectoderm GATA4 GATA4 GSC GATA4 T ISL1 NCAM1 FOXH1 NCAM1 MESP1 CER1 WNT3A MIXL1 GATA4 PAX6 CDX2 T PAX6 SOX17 HBB NES GATA6 WT1 SOX1 FN1 ACTC1 ZIC1 FOXA2 MYF5 ZIC1 CXCR4 TBX5 PAX6 NCAM1 TBX20 PAX6 KRT18 DDX4 TUBB3 EPCAM TBX5 SOX2 KRT18 NKX2-5 NES AFP COL1A1 +++ +++ 0 0 0 0 ++++ +++ ++++ +++ +++ ++++ +++ ++++ 0 0 0 0 +++ +++ ++++ +++ ++++ 0 0 0 0 hematopoietic potential H1 H1 H1 H1 H1 H1 PB6 PB6 PB7 PB7 PB6 PB6 PB7 PB6 PB6 PB6.1 PB6.1 PB6.1 PB6.1 PB6.1 PB6.1 PB12.1 PB12.1 PB12.1 PB12.1 PB12.1 PB12.1 Figure S2. Unsupervised hierarchical clustering of hPSC-derived EBs according to the mRNA expression of germ layer differentiation genes (microarray analysis) Selected ectoderm (A), endoderm (B) and mesoderm (C) related genes differentially expressed between hematopoietic-competent (H1, PB6.1, PB7) and -deficient cells (PB6, PB12.1) are shown (related to Table S1). -

Context-Dependent Roles in Cell and Tissue Physiology
Downloaded from http://cshperspectives.cshlp.org/ on September 24, 2021 - Published by Cold Spring Harbor Laboratory Press TGF-b and the TGF-b Family: Context-Dependent Roles in Cell and Tissue Physiology Masato Morikawa,1 Rik Derynck,2 and Kohei Miyazono3 1Ludwig Cancer Research, Science for Life Laboratory, Uppsala University, Biomedical Center, SE-751 24 Uppsala, Sweden 2Department of Cell and Tissue Biology, University of California at San Francisco, San Francisco, California 94143 3Department of Molecular Pathology, Graduate School of Medicine, The University of Tokyo, Bunkyo-ku, Tokyo 113-0033, Japan Correspondence: [email protected] The transforming growth factor-b (TGF-b) is the prototype of the TGF-b family of growth and differentiation factors, which is encoded by 33 genes in mammals and comprises homo- and heterodimers. This review introduces the reader to the TGF-b family with its complexity of names and biological activities. It also introduces TGF-b as the best-studied factor among the TGF-b family proteins, with its diversity of roles in the control of cell proliferation and differentiation, wound healing and immune system, and its key roles in pathology, for exam- ple, skeletal diseases, fibrosis, and cancer. lthough initially thought to stimulate cell TGF-b has been well documented in most cell Aproliferation, just like many growth factors, types, and has been best characterized in epithe- it became rapidly accepted that transforming lial cells. The bifunctional and context-depen- growth factor b (TGF-b) is a bifunctional reg- dent nature of TGF-b activities was further con- ulator that either inhibits or stimulates cell pro- firmed in a large variety of cell systems and liferation. -

BMP-13 Emerges As a Potential Inhibitor of Bone Formation
Short Research Communication BMP-13 Emerges as a Potential Inhibitor of Bone Formation Bojiang Shen1*, Divya Bhargav1, Aiqun Wei1, Lisa A Williams1, Helen Tao2, David D F Ma2 and Ashish D Diwan1 1. Orthopaedic Research Institute and Department of Orthopaedic Surgery, St George Hospital, University of New South Wales, Sydney, Australia 2. Blood Stem Cell and Cancer Research Unit, Department of Haematology, St. Vincent’s Hospital Sydney, University of New South Wales, Sydney, Australia * Correspondence to: Dr Bojiang Shen, Orthopaedic Research Institute, St George Clinical School, The University of New South Wales, 4-10 South Street, Kogarah, NSW 2217. Tel: 61-2-9113 2248; Fax: 61-2-9350 3967; E-mail: [email protected] Key words: BMP-13; GDF6; CDMP-2; osteogenic differentiation; mesenchymal stromal cells 1 ABSTRACT Bone morphogenetic protein-13 (BMP-13) plays an important role in skeletal development. In the light of a recent report that mutations in BMP-13 gene are associated with spine vertebral fusion in Klippel-Feil syndrome, we hypothesized that BMP-13 signaling is crucial for regulating embryonic endochondral ossification. In this study, we found that BMP-13 inhibited the osteogenic differentiation of human bone marrow multipotent mesenchymal stromal cells (BM MSCs) in vitro. The endogenous BMP13 gene expression in MSCs was examined under expansion conditions. The MSCs were then induced to differentiate into osteoblasts in osteo-inductive medium containing exogenous BMP-13. Gene expression was analysed by real-time PCR. Alkaline phosphatase (ALP) expression and activity, proteoglycan (PG) synthesis and matrix mineralization were assessed by cytological staining or ALP assay. -

Revised Figs 1-27-11 Nori
A TAC TAC TAC Cont 1wk 3wk 9wk pSmad2 tSmad2 Smad4 B AlexaFluor555-N-Ab DAPI CD68 Merged C TβR2FF TβR2cKD TβR1FF TβR1cKD TGFBR2 TGFBR1 GAPDH Supplemental Figure 1 A Ctgf B Ctgf * * 4 2 3 † 2 1 FoldIncrease 1 0 0 rhTGFβ1 - + - + rhTGFβ1 - + - + TβR2FF TβR2cKD DMSO Oxozeaenol C Nppa Nppb 1.6 * 2 * 1.2 1.6 1.2 0.8 0.8 FoldIncrease 0.4 0.4 0 0 rhTGFβ1 - + - + - + - + DMSO Oxozeaenol DMSO Oxozeaenol D rhTGFβ1 - + + Oxozeaenol - - + pp38 MAPK tp38 MAPK Supplemental Figure 2 A rhTGF 1 β 0 0 5 15 30 min TβR1FF pSmad3 tSmad3 pSmad3 TβR1cKD tSmad3 B TβR2FF TβR2cKD TβR1FF TβR1cKD TAC3wk TAC3wk TAC3w TAC3wk pSmad3 tSmad3 pTAK1 tTAK1 pMKK3/6 tMKK3 pp38MAPK tp38MAPK Supplemental Figure 3 Supplemental Table 1: Hemodynamic parameters and indices derived from PV loop analysis with N-Ab study at TAC-9wk Sham TAC+C-Ab TAC+N-Ab N=8 N=5 N=3 Parameter HR (/min) 533±11 486±9 514±15 LVPmax (mmHg) 105±3.8 174±3.8* 164±3.6* EDP (mmHg) 6.0±0.6 8.3±1.8 6.0±0.8 EDV (µL) 29.3±2.2 51.0±3.4* 75.7±13.4*† ESV (µL) 7.9±1.3 32.6±2.9* 60.6±12.5*† CO (mL/min) 11.4±0.6 8.8±0.5* 7.7±0.4* Ea (mmHg/uL) 4.5±0.3 9.4±1.0* 10.7±0.9* EF (%) 74.5±3.2 36.3±2.3* 20.7±2.5*† Systolic indexes Ees 8.4±0.7 6.8±1.6 6.5±2.2 PRSW 92.9±4.0 103.6±11.5* 76.3±10.0 dP/dtmax (mmHg/sec) 12951±701 9359±695 9627±308 PWRMax/EDV (mmHg/uL) 37.4±2.1 24.5±1.4* 14.4±1.9*† Diastolic indexes -dP/dtmin (mmHg/sec) -9555±694 -9709±674 -8078±290 Tau (msec) 4.2±0.24 5.1±0.26 6.7±0.5*† PFR/EDV (/sec) 30.6±2.4 16.0±3.0* 9.4±1.3* HR, heart rate; ESP, end-systolic pressure; EDP, end-diastolic pressure; EDV, end-diastolic -

GDF6 Gene Growth Differentiation Factor 6
GDF6 gene growth differentiation factor 6 Normal Function The GDF6 gene provides instructions for making a protein that is part of the transforming growth factor beta (TGFb ) superfamily, which is a group of proteins that help control the growth and development of tissues throughout the body. Within the TGFb superfamily, the GDF6 protein belongs to the bone morphogenetic protein family, which is involved in regulating the growth and maturation (differentiation) of bone and cartilage. Cartilage is a tough but flexible tissue that makes up much of the skeleton during early development. The proteins in this family are regulators of cell growth and differentiation both before and after birth. The GDF6 protein is necessary for the formation of bones and joints in the limbs, skull, spine, chest, and ribs. The protein is involved in setting up boundaries between bones during skeletal development. The GDF6 protein has also been found to be involved in the development of the eyes, specifically the specialized light-sensitive tissue that lines the back of the eye called the retina. The GDF6 protein likely plays a role in the survival of specialized cells within the retina that detect light and color (photoreceptor cells). Health Conditions Related to Genetic Changes Klippel-Feil syndrome At least 10 mutations in the GDF6 gene have been found to cause Klippel-Feil syndrome, a condition characterized by the abnormal joining (fusion) of two or more spinal bones in the neck (cervical vertebrae) and a variety of other features affecting many parts of the body. Most GDF6 gene mutations that cause Klippel-Feil syndrome replace single protein building blocks (amino acids) in the GDF6 protein. -
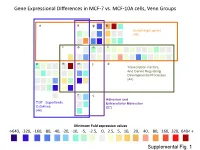
Supplementary Data
Gene Expressional Differences in MCF-7 vs. MCF-10A cells, Venn Groups e k g b Smad target genes (48) l o n i a h m j d Transcription Factors, And Genes Regulating Developmental Processes (44) f c Adhesion and TGF Superfamily Extracellular Molecules Cytokines (27) (44) Minimum Fold expression values ->640, -320, -160, -80, -40, -20, -10, -5, -2.5, 0, 2.5, 5, 10, 20, 40, 80, 160, 320, 640< + Supplemental Fig. 1 Supplemental Table 1. Change of Basal Gene Expressional values in MCF-7 as compared to MCF-10A cell line. Fold T-test Gene # GeneBank Symbol Up/Down p-Value Description /Position Regulation Venn Group a 01 /A01 NM_001105 ACVR1 -4.96 0.0000 Activin A receptor, type I 02 /A02 NM_001616 ACVR2A -2.62 0.0007 Activin A receptor, type IIA 03 /A03 NM_000020 ACVRL1 -1.25 0.7013 Activin A receptor type II-like 1 05 /A05 NM_020547 AMHR2 1.08 0.8043 Anti-Mullerian hormone receptor, type II 16 /B04 NM_004329 BMPR1A -2.40 0.0011 Bone morphogenetic protein receptor, type IA 17 /B05 NM_001203 BMPR1B -7.37 0.0000 Bone morphogenetic protein receptor, type IB 36 /C12 NM_000557 GDF5 -1.38 0.4911 Growth differentiation factor 5 (cartilage-derived morphogenetic protein-1) 37 /D01 NM_001001557 GDF6 1.12 0.9002 Growth differentiation factor 6 38 /D02 NM_182828 GDF7 -1.52 0.4995 Growth differentiation factor 7 53 /E05 NM_020997 LEFTY1 -1.76 0.0529 Left-right determination factor 1 59 /E11 NM_018055 NODAL -3.62 0.1290 Nodal homolog (mouse) 77 /G05 NM_003238 TGFB2 -4.52 0.0566 Transforming growth factor, beta 2 78 /G06 NM_003239 TGFB3 -1.12 0.2902 Transforming -

Recombinant Mouse GDF-7/BMP-12 Catalog Number: 779-G7/CF
Recombinant Mouse GDF-7/BMP-12 Catalog Number: 779-G7/CF DESCRIPTION Source E. coliderived Thr308Arg453 Accession # NP_038555 Nterminal Sequence Thr308 Analysis Structure / Form Disulfidelinked homodimer Predicted Molecular 15 kDa (monomer) Mass SPECIFICATIONS SDSPAGE 28 kDa, nonreducing conditions Activity Measured by its ability to induce alkaline phosphatase production by ATDC5 mouse chondrogenic cells. Nakamura, K. et al. (1999) Exp. Cell Res. 250:351. The ED50 for this effect is 0.251.25 µg/mL. Endotoxin Level <0.10 EU per 1 μg of the protein by the LAL method. Purity >95%, by SDSPAGE under reducing conditions and visualized by silver stain. Formulation Lyophilized from a 0.2 μm filtered solution in Acetonitrile and TFA. See Certificate of Analysis for details. PREPARATION AND STORAGE Reconstitution Reconstitute at 100 μg/mL in sterile 4 mM HCl. Shipping The product is shipped at ambient temperature. Upon receipt, store it immediately at the temperature recommended below. Stability & Storage Use a manual defrost freezer and avoid repeated freezethaw cycles. l 12 months from date of receipt, 20 to 70 °C as supplied. l 1 month, 2 to 8 °C under sterile conditions after reconstitution. l 3 months, 20 to 70 °C under sterile conditions after reconstitution. BACKGROUND Growth Differentiation Factor 7 (GDF7), also known as bone morphogenetic protein (BMP) 12 or cartilage derived morphogenetic protein 3 (CDMP3), is a member of the bone morphogenetic protein (BMP) family which belongs to the transforming growth factor β (TGFβ) superfamily. Mature GDF7 contains 146 amino acid (aa) residues with the characteristic seven conserved cysteine residues. -

CER1 Is a Common Target of WNT and NODAL Signaling Pathways in Human Embryonic Stem Cells
795-799 24/3/06 13:04 Page 795 INTERNATIONAL JOURNAL OF MOLECULAR MEDICINE 17: 795-799, 2006 795 CER1 is a common target of WNT and NODAL signaling pathways in human embryonic stem cells MASUKO KATOH1 and MASARU KATOH2 1M&M Medical BioInformatics, Hongo 113-0033; 2Genetics and Cell Biology Section, National Cancer Center Research Institute, Tokyo 104-0045, Japan Received January 3, 2006; Accepted February 7, 2006 Abstract. Nodal and BMP signaling pathways network with embryogenesis, underwent protein evolution as well as WNT signaling pathway during embryogenesis and carcino- promoter evolution. These facts indicate that molecular genesis. CER1 (Cerberus 1) and GREM3 (CKTSF1B3 or evolution of CER1 orthologs contributes to the significantly CER2) inhibit NODAL signaling through ACVR1B (ALK4) divergent scenarios of early embryogenesis in primates and or ACVR1C (ALK7) to SMAD2 or SMAD3. GREM1 rodents. (CKTSF1B1) inhibits BMP signaling through BMPR1A (ALK3), BMPR1B (ALK6) or ACVR1 (ALK2) to SMAD1, Introduction SMAD5 or SMAD8. CER1, GREM1 and GREM3 are DAN domain (DAND) family members; however, transcriptional TGFB1, TGFB2, TGFB3, NODAL, LEFTY1, LEFTY2, INHA, regulation of DAND family members by canonical WNT INHBA, INHBB, INHBC, INHBE, AMH, BMP2, BMP3, signaling pathway remains unclear. We searched for the BMP4, BMP5, BMP6, BMP7, BMP8A, BMP8B, BMP10, TCF/LEF-binding site within the promoter region of DAND BMP15, GDF1, GDF2, GDF3, GDF5, GDF6, GDF7, GDF8, family genes, including CER1, GREM1, GREM2, GREM3 and GDF9, GDF10, GDF11, and GDF15 are TGFß superfamily NBL1. Because triple TCF/LEF-binding sites were identified genes within the human genome (http://www.gene.ucl.ac.uk). within human CER1 promoter by using bioinformatics and TGFß signals are transduced through type I receptor TGFBR1 human intelligence, comparative genomics analyses on CER1 and type II receptor TGFBR2 to phosphorylate R-SMAD orthologs were further performed. -

Microrna-Regulated Protein-Protein Interaction Networks and Their Functions in Breast Cancer
Int. J. Mol. Sci. 2013, 14, 11560-11606; doi:10.3390/ijms140611560 OPEN ACCESS International Journal of Molecular Sciences ISSN 1422-0067 www.mdpi.com/journal/ijms Article MicroRNA-Regulated Protein-Protein Interaction Networks and Their Functions in Breast Cancer Chia-Hsien Lee 1,†, Wen-Hong Kuo 2,†, Chen-Ching Lin 1, Yen-Jen Oyang 1, Hsuan-Cheng Huang 3,* and Hsueh-Fen Juan 1,4,* 1 Graduate Institute of Biomedical Electronics and Bioinformatics, National Taiwan University, Taipei 106, Taiwan; E-Mails: [email protected] (C.-H.L.); [email protected] (C.-C.L.); [email protected] (Y.-J.O.) 2 Department of Physiology, College of Medicine, National Taiwan University, Taipei 100, Taiwan; E-Mail: [email protected] 3 Institute of Biomedical Informatics and Center for Systems and Synthetic Biology, National Yang-Ming University, Taipei 112, Taiwan 4 Institute of Molecular and Cellular Biology and Department of Life Science, National Taiwan University, Taipei 106, Taiwan † These authors contributed equally to this work. * Authors to whom correspondence should be addressed; E-Mails: [email protected] (H.-C.H.); [email protected] (H.-F.J.); Tel.: +886-2-2826-7357 (H.-C.H.); +886-2-3366-4536 (H.-F.J.); Fax: +886-2-2820-2508 (H.-C.H.); +886-2-2367-3374 (H.-F.J.). Received: 17 April 2013; in revised form: 21 May 2013 / Accepted: 22 May 2013 / Published: 30 May 2013 Abstract: MicroRNAs, which are small endogenous RNA regulators, have been associated with various types of cancer. Breast cancer is a major health threat for women worldwide. -

Bone Morphogenetic Proteins
Downloaded from http://cshperspectives.cshlp.org/ on September 24, 2021 - Published by Cold Spring Harbor Laboratory Press Bone Morphogenetic Proteins Takenobu Katagiri1 and Tetsuro Watabe2 1Division of Pathophysiology, Research Center for Genomic Medicine, Saitama Medical University, Hidaka-shi, Saitama 350-1241, Japan 2Section of Biochemistry, Department of Bio-Matrix, Graduate School of Medical and Dental Sciences, Tokyo Medical and Dental University, Bunkyo-ku, Tokyo 113-8549, Japan Correspondence: [email protected]; [email protected] Bone morphogenetic proteins (BMPs), originally identified as osteoinductive components in extracts derived from bone, are now known to play important roles in a wide array of pro- cesses during formation and maintenance of various organs including bone, cartilage, muscle, kidney, and blood vessels. BMPs and the related “growth and differentiation factors” (GDFs) are members of the transforming growth factor b (TGF-b) family, and trans- duce their signals through type I and type II serine–threonine kinase receptors and their intracellular downstream effectors, including Smad proteins. Furthermore, BMP signals are finely tuned by various agonists and antagonists. Because deregulation of the BMPactivity at multiple steps in signal transduction is linked to a wide variety of human diseases, therapeu- tic use of activators and inhibitors of BMP signaling will provide potential avenues for the treatment of the human disorders that are caused by hypo- and hyperactivation of BMP signals, respectively. he bone morphogenetic protein (BMP) (Senn 1889). In 1965, Urist reported that de- Tfamily of ligands plays important roles in a mineralized bone matrix implanted in muscular multitude of processes during embryonic devel- tissues induces ectopic formation of cartilage opment and adult homeostasis by regulating and bone tissues with bone marrow (Urist cellular lineage commitment, morphogenesis, 1965).