JUNB Regulatory Functions in Lymphangiogenesis: from Mescs to Zebrafish
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Activator Protein-1 Transcription Factor in Respiratory Epithelium Carcinogenesis
Subject Review The Activator Protein-1 Transcription Factor in Respiratory Epithelium Carcinogenesis Michalis V. Karamouzis,1 Panagiotis A. Konstantinopoulos,1,2 and Athanasios G. Papavassiliou1 1Department of Biological Chemistry, Medical School, University of Athens, Athens, Greece and 2Division of Hematology-Oncology, Beth Israel Deaconess Medical Center, Harvard Medical School, Boston, Massachusetts Abstract Much of the current anticancer research effort is focused on Respiratory epithelium cancers are the leading cause cell-surface receptors and their cognate upstream molecules of cancer-related death worldwide. The multistep natural because they provide the easiest route for drugs to affect history of carcinogenesis can be considered as a cellular behavior, whereas agents acting at the level of gradual accumulation of genetic and epigenetic transcription need to invade the nucleus. However, the aberrations, resulting in the deregulation of cellular therapeutic effect of surface receptor manipulation might be homeostasis. Growing evidence suggests that cross- considered less than specific because their actions are talk between membrane and nuclear receptor signaling modulated by complex interacting downstream signal trans- pathways along with the activator protein-1 (AP-1) duction pathways. A pivotal transcription factor during cascade and its cofactor network represent a pivotal respiratory epithelium carcinogenesis is activator protein-1 molecular circuitry participating directly or indirectly in (AP-1). AP-1–regulated genes include important modulators of respiratory epithelium carcinogenesis. The crucial role invasion and metastasis, proliferation, differentiation, and of AP-1 transcription factor renders it an appealing survival as well as genes associated with hypoxia and target of future nuclear-directed anticancer therapeutic angiogenesis (7). Nuclear-directed therapeutic strategies might and chemoprevention approaches. -

Transcriptional Control of Tissue-Resident Memory T Cell Generation
Transcriptional control of tissue-resident memory T cell generation Filip Cvetkovski Submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Graduate School of Arts and Sciences COLUMBIA UNIVERSITY 2019 © 2019 Filip Cvetkovski All rights reserved ABSTRACT Transcriptional control of tissue-resident memory T cell generation Filip Cvetkovski Tissue-resident memory T cells (TRM) are a non-circulating subset of memory that are maintained at sites of pathogen entry and mediate optimal protection against reinfection. Lung TRM can be generated in response to respiratory infection or vaccination, however, the molecular pathways involved in CD4+TRM establishment have not been defined. Here, we performed transcriptional profiling of influenza-specific lung CD4+TRM following influenza infection to identify pathways implicated in CD4+TRM generation and homeostasis. Lung CD4+TRM displayed a unique transcriptional profile distinct from spleen memory, including up-regulation of a gene network induced by the transcription factor IRF4, a known regulator of effector T cell differentiation. In addition, the gene expression profile of lung CD4+TRM was enriched in gene sets previously described in tissue-resident regulatory T cells. Up-regulation of immunomodulatory molecules such as CTLA-4, PD-1, and ICOS, suggested a potential regulatory role for CD4+TRM in tissues. Using loss-of-function genetic experiments in mice, we demonstrate that IRF4 is required for the generation of lung-localized pathogen-specific effector CD4+T cells during acute influenza infection. Influenza-specific IRF4−/− T cells failed to fully express CD44, and maintained high levels of CD62L compared to wild type, suggesting a defect in complete differentiation into lung-tropic effector T cells. -

Junb and GATA3 Transcription Factors Cytokine Expression Through Regulating CARMA1 Controls Th2 Cell-Specific
CARMA1 Controls Th2 Cell-Specific Cytokine Expression through Regulating JunB and GATA3 Transcription Factors This information is current as Marzenna Blonska, Donghyun Joo, Patrick A. of September 27, 2021. Zweidler-McKay, Qingyu Zhao and Xin Lin J Immunol published online 27 February 2012 http://www.jimmunol.org/content/early/2012/02/26/jimmun ol.1102943 Downloaded from Supplementary http://www.jimmunol.org/content/suppl/2012/02/27/jimmunol.110294 Material 3.DC1 http://www.jimmunol.org/ Why The JI? Submit online. • Rapid Reviews! 30 days* from submission to initial decision • No Triage! Every submission reviewed by practicing scientists • Fast Publication! 4 weeks from acceptance to publication by guest on September 27, 2021 *average Subscription Information about subscribing to The Journal of Immunology is online at: http://jimmunol.org/subscription Permissions Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Email Alerts Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts The Journal of Immunology is published twice each month by The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2012 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. Published February 27, 2012, doi:10.4049/jimmunol.1102943 The Journal of Immunology CARMA1 Controls Th2 Cell-Specific Cytokine Expression through Regulating JunB and GATA3 Transcription Factors Marzenna Blonska,* Donghyun Joo,*,† Patrick A. Zweidler-McKay,‡ Qingyu Zhao,*,1 and Xin Lin*,† The scaffold protein CARMA1 is required for the TCR-induced lymphocyte activation. -

Junb Differs from C-Jun in Its DNA- Binding and Dlmenzatlon Domains, and Represses C-Jun by Formation of Inactive Heterodimers
Downloaded from genesdev.cshlp.org on October 3, 2021 - Published by Cold Spring Harbor Laboratory Press JunB differs from c-Jun in its DNA- binding and dlmenzatlon domains, and represses c-Jun by formation of inactive heterodimers Tiliang Deng and Michael Karin 1 Department of Pharmacology, Center for Molecular Genetics, University of California, San Diego, School of Medicine, La Jolla, California 92093-0636 USA JunB differs considerably from c-Jun in its ability to activate AP-l-responsive genes and induce oncogenic transformation. We demonstrate that the decreased ability of JunB to activate gene expression is the result of a small number of amino acid changes between its DNA-binding and dimerization motifs and the corresponding regions of c-Jun. These changes lead to a 10-fold decrease in the DNA-binding activity of Junk JunB can be converted into a c-Jun-like activator by substituting four amino acids in its DNA-binding and dimerization motifs with the corresponding c-Jun sequences. JunB can also attenuate trans-activation by c-Jun, an activity mediated by its leucine zipper. This ability depends on two glycine residues that decrease the stability of the JunB leucine zipper, resulting in decreased homodimerization and increased heterodimerization. These results illustrate how small changes in primary structure, including chemically conservative changes, can result in functional divergence of two highly related transcriptional regulators. [Key Words: JunB; c-Jun; DNA-binding/dimerization motifs; leucine zipper] Received October 16, 1992; revised version accepted December 29, 1992. Transcriptional control is commonly mediated by se- AP-1 was identified as a transcription factor required quence-specific DNA-binding proteins (Pabo and Saner for optimal activity of the human metallothionein IIA 1984; Johnson and McKnight 1989). -

1714 Gene Comprehensive Cancer Panel Enriched for Clinically Actionable Genes with Additional Biologically Relevant Genes 400-500X Average Coverage on Tumor
xO GENE PANEL 1714 gene comprehensive cancer panel enriched for clinically actionable genes with additional biologically relevant genes 400-500x average coverage on tumor Genes A-C Genes D-F Genes G-I Genes J-L AATK ATAD2B BTG1 CDH7 CREM DACH1 EPHA1 FES G6PC3 HGF IL18RAP JADE1 LMO1 ABCA1 ATF1 BTG2 CDK1 CRHR1 DACH2 EPHA2 FEV G6PD HIF1A IL1R1 JAK1 LMO2 ABCB1 ATM BTG3 CDK10 CRK DAXX EPHA3 FGF1 GAB1 HIF1AN IL1R2 JAK2 LMO7 ABCB11 ATR BTK CDK11A CRKL DBH EPHA4 FGF10 GAB2 HIST1H1E IL1RAP JAK3 LMTK2 ABCB4 ATRX BTRC CDK11B CRLF2 DCC EPHA5 FGF11 GABPA HIST1H3B IL20RA JARID2 LMTK3 ABCC1 AURKA BUB1 CDK12 CRTC1 DCUN1D1 EPHA6 FGF12 GALNT12 HIST1H4E IL20RB JAZF1 LPHN2 ABCC2 AURKB BUB1B CDK13 CRTC2 DCUN1D2 EPHA7 FGF13 GATA1 HLA-A IL21R JMJD1C LPHN3 ABCG1 AURKC BUB3 CDK14 CRTC3 DDB2 EPHA8 FGF14 GATA2 HLA-B IL22RA1 JMJD4 LPP ABCG2 AXIN1 C11orf30 CDK15 CSF1 DDIT3 EPHB1 FGF16 GATA3 HLF IL22RA2 JMJD6 LRP1B ABI1 AXIN2 CACNA1C CDK16 CSF1R DDR1 EPHB2 FGF17 GATA5 HLTF IL23R JMJD7 LRP5 ABL1 AXL CACNA1S CDK17 CSF2RA DDR2 EPHB3 FGF18 GATA6 HMGA1 IL2RA JMJD8 LRP6 ABL2 B2M CACNB2 CDK18 CSF2RB DDX3X EPHB4 FGF19 GDNF HMGA2 IL2RB JUN LRRK2 ACE BABAM1 CADM2 CDK19 CSF3R DDX5 EPHB6 FGF2 GFI1 HMGCR IL2RG JUNB LSM1 ACSL6 BACH1 CALR CDK2 CSK DDX6 EPOR FGF20 GFI1B HNF1A IL3 JUND LTK ACTA2 BACH2 CAMTA1 CDK20 CSNK1D DEK ERBB2 FGF21 GFRA4 HNF1B IL3RA JUP LYL1 ACTC1 BAG4 CAPRIN2 CDK3 CSNK1E DHFR ERBB3 FGF22 GGCX HNRNPA3 IL4R KAT2A LYN ACVR1 BAI3 CARD10 CDK4 CTCF DHH ERBB4 FGF23 GHR HOXA10 IL5RA KAT2B LZTR1 ACVR1B BAP1 CARD11 CDK5 CTCFL DIAPH1 ERCC1 FGF3 GID4 HOXA11 IL6R KAT5 ACVR2A -

The Retinoblastoma Gene Family Members Prb and P107 Coactivate the AP-1-Dependent Mouse Tissue Factor Promoter in ®Broblasts
Oncogene (2000) 19, 3352 ± 3362 ã 2000 Macmillan Publishers Ltd All rights reserved 0950 ± 9232/00 $15.00 www.nature.com/onc The retinoblastoma gene family members pRB and p107 coactivate the AP-1-dependent mouse tissue factor promoter in ®broblasts Su-Ling Liu1, Arlymae Rand1, Robert J Kelm Jr,*,1 and Michael J Getz1,2 1Department of Biochemistry and Molecular Biology, Program in Tumor Biology, Mayo Clinic/Foundation, Rochester, Minnesota, MN 55905, USA Serum-stimulation of quiescent mouse ®broblasts results al., 1996), to maintenance of the placental labyrinth in transcriptional activation of tissue factor (TF), the during gestation (Erlich et al., 1999), and to tumor cellular initiator of blood coagulation. This requires the angiogenesis (Contrino et al., 1996). Tissue factor has rapid entry of c-Fos into speci®c AP-1 DNA-binding also been implicated as a determinant of metastatic complexes and can be strongly inhibited by the potential in melanoma cells (Mueller et al., 1992; adenovirus E1A 12S gene product. In this study, we Bromberg et al., 1995) and expression of TF in the utilized a panel of E1A mutants de®cient in cellular stromal compartment of breast carcinomas has been protein binding to analyse the molecular basis for E1A shown to correlate with progression to invasive inhibition of a minimal, c-Fos-dependent TF promoter/ cancer (Contrino et al., 1996; Vrana et al., 1996). reporter construct in mouse AKR-2B ®broblasts. Muta- Thus, it is important to understand how TF gene tions which impaired binding of the retinoblastoma tumor expression is regulated in both normal and neoplastic suppressor protein family members pRB, p107, and p130 cell types. -

Alterations of NFIA in Chronic Malignant Myeloid Diseases
Letters to the Editor 583 BACH2 is a B-cell specific transcription factor that induces 2Laboratoire d0He´matologie, CHU de Caen, Caen, France; 3 apoptosis in lymphoma cells and reduced expression has been De´parement de Genetique, Laboratoire Pasteur, Cerba, France and associated with poor prognostic outcome in diffuse large B-cell 4 lymphoma.7 Department of Haematology, John Radcliffe Hospital, Oxford, UK Our data also suggested that many components of the E-mail: [email protected] transforming growth factor-b (TGFb) signalling pathway are downregulated in PPBL (Supplementary Figure S3), including the tumour-suppressor molecule SMAD4 (Figure 1i; P ¼ 0.007). The TGFb pathway negatively regulates cell growth, and References inactivation of SMAD4 is present in over half of pancreatic 8 cancers and up to 35% of colorectal cancers. This is as far as 1 Mossafa H, Malaure H, Maynadie M, Valensi F, Schillinger F, we know the first description of the downregulation of SMAD4 Garand R et al. Persistent polyclonal B lymphocytosis with in a B-cell disorder. binucleated lymphocytes: a study of 25 cases. Groupe Francais PPBL represents a useful model for B-cell dysfunction as it d’Hematologie Cellulaire. Br J Haematol 1999; 104: 486–493. displays characteristics associated with benign (polyclonality, 2 Ozanne BW, Spence HJ, McGarry LC, Hennigan RF. Transcription factors control invasion: AP-1 the first among equals. Oncogene benign clinical course) and malignant disease (frequent chromo- 2007; 26: 1–10. somal abnormalities and aberrant B-cell morphology). Nearly all 3 Watanabe M, Sasaki M, Itoh K, Higashihara M, Umezawa K, Kadin studies to date have sought to characterize the morphological or ME et al. -

The Tumor Suppressor HHEX Inhibits Axon Growth When Prematurely Expressed in Developing Central Nervous System Neurons
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by epublications@Marquette Marquette University e-Publications@Marquette Biological Sciences Faculty Research and Biological Sciences, Department of Publications 9-1-2015 The umorT Suppressor HHEX Inhibits Axon Growth when Prematurely Expressed in Developing Central Nervous System Neurons Matthew .T Simpson Marquette University Ishwariya Venkatesh Marquette University Ben L. Callif Marquette University Laura K. Thiel Marquette University Denise M. Coley Marquette University See next page for additional authors Accepted version. Molecular and Cellular Neuroscience, Vol 68 )September 2015): 272-283. DOI. © 2015 Elsevier Inc. Used with permission. NOTICE: this is the author’s version of a work that was accepted for publication in Molecular and Cellular Neuroscience. Changes resulting from the publishing process, such as peer review, editing, corrections, structural formatting, and other quality control mechanisms may not be reflected in this document. Changes may have been made to this work since it was submitted for publication. A definitive version was subsequently published in Molecular and Cellular Neuroscience, Vol 68 )September 2015): 272-283. DOI. Authors Matthew T. Simpson, Ishwariya Venkatesh, Ben L. Callif, Laura K. Thiel, Denise M. Coley, Kristen N. Winsor, Zimei Wang, Audra A. Kramer, Jessica K. Lerch, and Murray G. Blackmore This article is available at e-Publications@Marquette: https://epublications.marquette.edu/bio_fac/515 NOT THE PUBLISHED VERSION; this is the author’s final, peer-reviewed manuscript. The published version may be accessed by following the link in the citation at the bottom of the page. The Tumor Suppressor HHEX Inhibits Axon Growth When Prematurely Expressed in Developing Central Nervous System Neurons Matthew T. -

Transcription Factors Recognize DNA Shape Without Nucleotide Recognition
bioRxiv preprint doi: https://doi.org/10.1101/143677; this version posted May 29, 2017. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Transcription+factors+recognize+DNA+shape+without+nucleotide+recognition++ Md.$Abul$Hassan$Samee1,$Benoit$G.$Bruneau1,2,$Katherine$S.$Pollard$1,3$ 1:$Gladstone$Institutes,$1650$Owens$St.,$San$Francisco,$CA$94158$ 2:$$Department$of$Pediatrics$and$Cardiovascular$Research$Institute,$University$of$California,$ San$Francisco,$CA$94158$ 3:$Division$of$Bioinformatics,$Institute$for$Human$Genetics,$and$Institute$for$Computational$ Health$Sciences,$University$of$California,$San$Francisco,$CA$94158$ $ + bioRxiv preprint doi: https://doi.org/10.1101/143677; this version posted May 29, 2017. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Abstract+ We$hypothesized$that$transcription$factors$(TFs)$recognize$DNA$shape$without$nucleotide$ sequence$recognition.$Motivating$an$independent$role$for$shape,$many$TF$binding$sites$lack$ a$sequenceZmotif,$DNA$shape$adds$specificity$to$sequenceZmotifs,$and$different$sequences$ can$ encode$ similar$ shapes.$ We$ therefore$ asked$ if$ binding$ sites$ of$ a$ TF$ are$ enriched$ for$ specific$patterns$of$DNA$shapeZfeatures,$e.g.,$helical$twist.$We$developed$ShapeMF,$which$ discovers$ these$ shapeZmotifs$ de%novo%without$ taking$ sequence$ information$ into$ account.$ -

Comprehensive Epigenomic Profiling of Human Alveolar Epithelial
bioRxiv preprint doi: https://doi.org/10.1101/2021.04.14.439786; this version posted April 15, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. FULL TITLE: Comprehensive epigenomic profiling of human alveolar epithelial differentiation identifies key epigenetic states and transcription factor co-regulatory networks for maintenance of distal lung identity SHORT TITLE: Epigenomic profiling of human alveolar epithelial differentiation Beiyun Zhou1,2,3, Theresa Ryan Stueve3,4,5, Evan A. Mihalakakos3,4,5, Lin Miao3,4,5, Daniel J Mullen3,4,5, Yinchong Wang3,4,5, Yixin Liu1, Jiao Luo1, Evelyn Tran3,4,5, Kim D. Siegmund6, Sean K. Lynch7, Amy L. Ryan1,2,8, Ite A. Offringa3,4,5,#, Zea Borok1,2,3,5,9,#, Crystal N. Marconett3,4,5* 1Division of Pulmonary, Critical Care and Sleep Medicine, Department of Medicine, 2Hastings Center for Pulmonary Research, 3Norris Comprehensive Cancer Center, Departments of 4Surgery, 5Biochemistry and Molecular Medicine, and 6Preventive Medicine, Keck School of Medicine, University of Southern California, USA 90089 7Department of Engineering, Test Manufacturing Group, MAXIM Integrated Products, Sunnyvale, California, USA 95134 8Department of Stem Cell Biology and Regenerative Medicine, University of Southern California, Los Angeles, CA USA 90089 9Division of Pulmonary, Critical Care and Sleep Medicine, Department of Medicine, University of California, San Diego, La Jolla, CA USA 92093. #These authors contributed equally * Corresponding/Senior author contact information: Crystal Marconett, Ph.D., Norris Comprehensive Cancer Center, Keck School of Medicine, University of Southern California, NTT 6418A M/C 9176, Los Angeles CA 90089-9520, USA. -
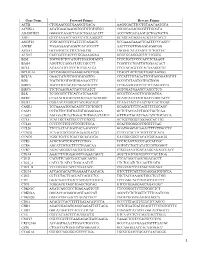
Gene Name Forward Primer Reverse Primer ACTB
Gene Name Forward Primer Reverse Primer ACTB CTGGAACGGTGAAGGTGACA AAGGGACTTCCTGTAACAATGCA ACVRL1 ACATGAAGAAGGTGGTGTGTGTGG CGGGCAGAGGGGTTTGGGTA ADAMDEC1 GGGGCCAGACTACACTGAAACATT ACCCGTCACAAGTACTGATGCTG AHI1 GTCCAAAACTACCCCATCAAGGCT GCAGCACAGGAACGTATCACCT ANGPT2 TGGCAGCGTTGATTTTCAGAGG GCGAAACAAACTCATTTCCCAGCC ANPEP TGAAGAAGCAGGTCACACCCCT AACTCCGTTGGAGCAGGCGG APOA1 GCCGTGCTCTTCCTGACGG TGGGACACATAGTCTCTGCCGC ATXN7 CACCGCCCACTCTGGAAAAGAA GGGTGCAGGGCTTCTTGGTG B2M TGCTGTCTCCATGTTTGATGTATCT TCTCTGCTCCCCACCTCTAAGT BAG4 AGGTTCCAGGATATCCGCCTT TCGGTCCTGATTGTGGAACACT BCL2 ACAACATCGCCCTGTGGATGA CCGTACAGTTCCACAAAGGCAT BCL2L14 GCTCAGGGTCAAAGGACGTTGG TCAGCTACTCGGTTGGCAATGG BCL7A GAACCATGTCGGGCAGGTCG CCCATTTGTAGATTCGTAGGGATGTGT BIN1 TGCTGTCGTGGTGGAGACCTTC GCCGTGTAGTCGTGCTGGG BIRC3 TGCTATCCACATCAGACAGCCC TCTGAATGGTCTTCTCCAGGTTCA BIRC5 TTCTCAAGGACCACCGCATCT AGTGGATGAAGCCAGCCTCG BLK TCGGGGTCTTCACCATCAAAGC GCGCTCCAGGTTGCGGATGA BTRC CCAAATGTGTCATTACCAACATGGGC GCAGCACATAGTGATTTGGCATCC BUB3 CGGAACATGGGTTACGTGCAGC CCAAATACTCAACTGCCACTCGGC CAGE1 TCCAAAATGCACAGTCTTCTGGCT GGAGGCTCTTCAGTTTTTGCAGC CASP1 CCTGTTCCTGTGATGTGGAGGAAA GCTCTACCATCTGGCTGCTCAA CASP3 AGCGAATCAATGGACTCTGGAATATCC GTTTGCTGCATCGACATCTGTACCA CCL5 TCATTGCTACTGCCCTCTGCG ACTGCTGGGTTGGAGCACTTG CCL18 CCCTCCTTGTCCTCGTCTGCA GCACTGGGGGCTGGTTTCAG CCL26 TTCCAATACAGCCACAAGCCCC GGATGGGTACAGACTTTCTTGCCTC CCND2 TCAAGTGCGTGCAGAAGGACAT CTTCGCACTTCTGTTCCTCACA CCND3 TGGCTGCTGTGATTGCACATGA GATGGCGGGTACATGGCAAAGG CCR3 ACGCTGCTCTGCTTCCTGG TCCTCAGTTCCCCACCATCGC CCR4 AGCATCGTGCTTCCTGAGCAA GGTGTCTGCTATATCCGTGGGGT CCR7 AGACAGGGGTAGTGCGAGGC -

Xo PANEL DNA GENE LIST
xO PANEL DNA GENE LIST ~1700 gene comprehensive cancer panel enriched for clinically actionable genes with additional biologically relevant genes (at 400 -500x average coverage on tumor) Genes A-C Genes D-F Genes G-I Genes J-L AATK ATAD2B BTG1 CDH7 CREM DACH1 EPHA1 FES G6PC3 HGF IL18RAP JADE1 LMO1 ABCA1 ATF1 BTG2 CDK1 CRHR1 DACH2 EPHA2 FEV G6PD HIF1A IL1R1 JAK1 LMO2 ABCB1 ATM BTG3 CDK10 CRK DAXX EPHA3 FGF1 GAB1 HIF1AN IL1R2 JAK2 LMO7 ABCB11 ATR BTK CDK11A CRKL DBH EPHA4 FGF10 GAB2 HIST1H1E IL1RAP JAK3 LMTK2 ABCB4 ATRX BTRC CDK11B CRLF2 DCC EPHA5 FGF11 GABPA HIST1H3B IL20RA JARID2 LMTK3 ABCC1 AURKA BUB1 CDK12 CRTC1 DCUN1D1 EPHA6 FGF12 GALNT12 HIST1H4E IL20RB JAZF1 LPHN2 ABCC2 AURKB BUB1B CDK13 CRTC2 DCUN1D2 EPHA7 FGF13 GATA1 HLA-A IL21R JMJD1C LPHN3 ABCG1 AURKC BUB3 CDK14 CRTC3 DDB2 EPHA8 FGF14 GATA2 HLA-B IL22RA1 JMJD4 LPP ABCG2 AXIN1 C11orf30 CDK15 CSF1 DDIT3 EPHB1 FGF16 GATA3 HLF IL22RA2 JMJD6 LRP1B ABI1 AXIN2 CACNA1C CDK16 CSF1R DDR1 EPHB2 FGF17 GATA5 HLTF IL23R JMJD7 LRP5 ABL1 AXL CACNA1S CDK17 CSF2RA DDR2 EPHB3 FGF18 GATA6 HMGA1 IL2RA JMJD8 LRP6 ABL2 B2M CACNB2 CDK18 CSF2RB DDX3X EPHB4 FGF19 GDNF HMGA2 IL2RB JUN LRRK2 ACE BABAM1 CADM2 CDK19 CSF3R DDX5 EPHB6 FGF2 GFI1 HMGCR IL2RG JUNB LSM1 ACSL6 BACH1 CALR CDK2 CSK DDX6 EPOR FGF20 GFI1B HNF1A IL3 JUND LTK ACTA2 BACH2 CAMTA1 CDK20 CSNK1D DEK ERBB2 FGF21 GFRA4 HNF1B IL3RA JUP LYL1 ACTC1 BAG4 CAPRIN2 CDK3 CSNK1E DHFR ERBB3 FGF22 GGCX HNRNPA3 IL4R KAT2A LYN ACVR1 BAI3 CARD10 CDK4 CTCF DHH ERBB4 FGF23 GHR HOXA10 IL5RA KAT2B LZTR1 ACVR1B BAP1 CARD11 CDK5 CTCFL DIAPH1 ERCC1 FGF3 GID4 HOXA11