Atomic and Ionic Radii of Elements 1–96 Martinrahm,*[A] Roald Hoffmann,*[A] and N
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
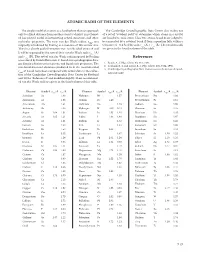
ATOMIC RADII of the ELEMENTS References
ATOMIC RADII OF THE ElEMENTS The simple model of an atom as a hard sphere that can approach The Cambridge Crystallographic Data Center also makes use only to a fixed distance from another atom to which it is not bond- of a set of “covalent radii” to determine which atoms in a crystal ed has proved useful in interpreting crystal structures and other are bonded to each other . Thus two atoms A and B are judged to molecular properties . The term van der Waals radius, rvdw, was be connected by a covalent bond if their separation falls within a originally introduced by Pauling as a measure of this atomic size . tolerance of ±0 .4 Å of the sum rcov (A) + rcov (B) . The covalent radii Thus in a closely packed structure two non-bonded atoms A and are given in the fourth column of the table . B will be separated by the sum of their van der Waals radii rvdw (A) and rvdw (B) . The set of van der Waals radii proposed by Pauling References was refined by Bondi (Reference 1) based on crystallographic data, gas kinetic collision cross sections, and liquid state properties . The 1 . Bondi, A ., J. Phys. Chem. 68, 441, 1964 . non-bonded contact distances predicted from the recommended 2 . Rowland, R . S . and Taylor, R ., J. Phys. Chem. 100, 7384, 1996 . 3 . Cambridge Crystallographic Data Center, www .ccdc .cam .ac .uk/prod- r of Bondi have been compared with actual data in the collec- vdw ucts/csd/radii/ tion of the Cambridge Crystallographic Data Center by Rowland and Taylor (Reference 2) and modified slightly . -

Modeling the Shape of Ions in Pyrite-Type Crystals
Crystals 2014, 4, 390-403; doi:10.3390/cryst4030390 OPEN ACCESS crystals ISSN 2073-4352 www.mdpi.com/journal/crystals Article Modeling the Shape of Ions in Pyrite-Type Crystals Mario Birkholz IHP, Im Technologiepark 25, 15236 Frankfurt (Oder), Germany; E-Mail: [email protected]; Tel.: +49-335-56250 Received: 13 April 2014; in revised form: 22 August 2014 / Accepted: 26 August 2014 / Published: 3 September 2014 Abstract: The geometrical shape of ions in crystals and the concept of ionic radii are re-considered. The re-investigation is motivated by the fact that a spherical modelling is justified for p valence shell ions on cubic lattice sites only. For the majority of point groups, however, the ionic radius must be assumed to be an anisotropic quantity. An appropriate modelling of p valence ions then has to be performed by ellipsoids. The approach is tested for pyrite-structured dichalcogenides MX2, with chalcogen ions X = O, S, Se and Te. The latter are found to exhibit the shape of ellipsoids being compressed along the <111> symmetry axes, with two radii r|| and describing their spatial extension. Based on this ansatz, accurate interatomic M–X distances can be derived and a consistent geometrical model emerges for pyrite-structured compounds. Remarkably, the volumes of chalcogen ions are found to vary only little in different MX2 compounds, suggesting the ionic volume rather than the ionic radius to behave as a crystal-chemical constant. Keywords: ionic radius; ionic shape; bonding distance; ionic volume; pyrite-type compounds; di-chalcogenides; di-oxides; di-sulfides; di-selenides; di-tellurides 1. -

A Study of the Hydration of the Alkali Metal Ions in Aqueous Solution
Article pubs.acs.org/IC A Study of the Hydration of the Alkali Metal Ions in Aqueous Solution Johan Mahler̈ and Ingmar Persson* Department of Chemistry, Swedish University of Agricultural Sciences, P.O. Box 7015, SE-750 07 Uppsala, Sweden *S Supporting Information ABSTRACT: The hydration of the alkali metal ions in aqueous solution has been studied by large angle X-ray scattering (LAXS) and double difference infrared spectroscopy (DDIR). The structures of the dimethyl sulfoxide solvated alkali metal ions in solution have been determined to support the studies in aqueous solution. The results of the LAXS and DDIR mea- surements show that the sodium, potassium, rubidium and cesium ions all are weakly hydrated with only a single shell of water molecules. The smaller lithium ion is more strongly hydrated, most probably with a second hydration shell present. The influence of the rubidium and cesium ions on the water structure was found to be very weak, and it was not possible to quantify this effect in a reliable way due to insufficient separation of the O−D stretching bands of partially deuterated water bound to these metal ions and the O−D stretching bands of the bulk water. Aqueous solutions of sodium, potassium and cesium iodide and cesium and lithium hydroxide have been studied by LAXS and M−O bond distances have been determined fairly accurately except for lithium. However, the number of water molecules binding to the alkali metal ions is very difficult to determine from the LAXS measurements as the number of distances and the temperature factor are strongly correlated. -

ARC: an Open-Source Library for Calculating Properties of Alkali Rydberg Atoms
ARC: An open-source library for calculating properties of alkali Rydberg atoms N. Šibalic´a,∗, J. D. Pritchardb, C. S. Adamsa, K. J. Weatherilla aJoint Quantum Center (JQC) Durham-Newcastle, Department of Physics, Durham University, South Road, Durham, DH1 3LE, United Kingdom bDepartment of Physics, SUPA, University of Strathclyde, 107 Rottenrow East, Glasgow, G4 0NG, United Kingdom Abstract We present an object-oriented Python library for computation of properties of highly-excited Rydberg states of alkali atoms. These include single-body effects such as dipole matrix elements, excited-state lifetimes (radiative and black-body limited) and Stark maps of atoms in external electric fields, as well as two-atom interaction potentials accounting for dipole and quadrupole coupling effects valid at both long and short range for arbitrary placement of the atomic dipoles. The package is cross-referenced to precise measurements of atomic energy levels and features extensive documentation to facilitate rapid upgrade or expansion by users. This library has direct application in the field of quantum information and quantum optics which exploit the strong Rydberg dipolar interactions for two-qubit gates, robust atom-light interfaces and simulating quantum many-body physics, as well as the field of metrology using Rydberg atoms as precise microwave electrometers. Keywords: Alkali atom, Matrix elements, Dipole-dipole interactions, Stark shift, Förster resonances PROGRAM SUMMARY They are a flourishing field for quantum information process- Program Title: ARC: Alkali Rydberg Calculator ing [1, 2] and quantum optics [3, 4, 5] in the few to single exci- Licensing provisions: BSD-3-Clause tation regime, as well as many-body physics [6, 7, 8, 9, 10, 11], Programming language: Python 2.7 or 3.5, with C extension in the many-excitations limit. -

Chalcogen-Nitrogen Bond: Insights Into a Key Chemical Motif
Proceedings Chalcogen-nitrogen Bond: Insights into A Key Chemical Motif Marco Bortoli,1 Andrea Madabeni,1 Pablo Andrei Nogara,2 Folorunsho B. Omage,2 Giovanni Ribaudo,3 Davide Zeppilli,1 Joao Batista Teixeira Rocha,2* Laura Orian1* 1 Dipartimento di Scienze Chimiche Università degli Studi di Padova Via Marzolo 1 35131 Padova, Italy; [email protected] (M.B.); [email protected] (A.M..); [email protected] (D.Z.) 2 Departamento de Bioquímica e Biologia Molecular, Universidade Federal de Santa Maria, Santa Maria, 97105-900, RS Brazil; [email protected] (P.A.N.); [email protected] (F.B.O.) 3 Dipartimento di Medicina Molecolare e Traslazionale, Università degli Studi di Brescia, Viale Europa 11, 25123 Brescia, Italy; [email protected] (G.R.) * Correspondence: : [email protected] (J.B.T.R), [email protected] (L.O.); † Presented at the 1st International Electronic Conference on Catalysis Sciences, 10–30 November 2020; Available online: https://eccs2020.sciforum.net/ Published: 10 November 2020 Abstract: Chalcogen-nitrogen chemistry deals with systems in which sulfur, selenium or tellurium is linked to a nitrogen nucleus. This chemical motif is a key component of different functional structures, ranging from inorganic materials and polymers to rationally designed catalysts, to bioinspired molecules and enzymes. The formation of a selenium-nitrogen bond, and its disruption, are rather common events in organic Se-catalyzed processes. In nature, along the mechanistic path of glutathione peroxidase, evidence of the formation of a Se-N bond in highly oxidizing conditions has been reported and interpreted as a strategy to protect the selenoenzyme from overoxidation. -

Van Der Waals Radii of Elements S
Inorganic Materials, Vol. 37, No. 9, 2001, pp. 871–885. Translated from Neorganicheskie Materialy, Vol. 37, No. 9, 2001, pp. 1031–1046. Original Russian Text Copyright © 2001 by Batsanov. Van der Waals Radii of Elements S. S. Batsanov Center for High Dynamic Pressures, Mendeleevo, Solnechnogorskii raion, Moscow oblast, 141570 Russia Received February 14, 2001 Abstract—The available data on the van der Waals radii of atoms in molecules and crystals are summarized. The nature of the continuous variation in interatomic distances from van der Waals to covalent values and the mechanisms of transformations between these types of chemical bonding are discussed. INTRODUCTION der Waals radius with the quantum-mechanical require- ment that the electron density vary continuously at the The notion that an interatomic distance can be periphery of atoms. thought of as the sum of atomic radii was among the most important generalizations in structural chemistry, In this review, the van der Waals radii of atoms eval- treating crystals and molecules as systems of interact- uated from XRD data, molar volumes, physical proper- ing atoms (Bragg, 1920). The next step forward in this ties, and crystal-chemical considerations are used to area was taken by Mack [1] and Magat [2], who intro- develop a universal system of van der Waals radii. duced the concept of nonvalent radius (R) for an atom situated at the periphery of a molecule and called it the atomic domain radius [1] or Wirkungsradius [2], ISOTROPIC CRYSTALLOGRAPHIC implying that this radius determines intermolecular dis- VAN DER WAALS RADII tances. Later, Pauling [3] proposed to call it the van der Kitaigorodskii [4, 5] was the first to formulate the Waals radius, because it characterizes van der Waals principle of close packing of molecules in crystalline interactions between atoms. -

Lawrence Berkeley Laboratory· UNIVERSITY of CALIFORNIA'
LBL-37435 UC-800 Lawrence Berkeley Laboratory· UNIVERSITY OF CALIFORNIA' To be published as a chapter in Frontiers in Nuclear Chemistry, D.D. Sood, Ed., Indian Association of Nuclear Chemists and Allied Scientists, Bombay, India, 1995 Summary of the Properties of the Lanthanide and Actinide Elements G.T. Seaborg and D.E. Hobart June 1995 --- :0 I'T1 ..(") em'"T1 -,o::c oCDm S::III:Z _. (") QI:ZI'T1 r+~(") CD 0 "'0 CD_. -< c.--- CQ. r t:Dr- 1 w...... ~ w U1 Prepared for the U.S. Department of Energy under Contract Number DE-AC03-76SF00098 DISCLAIMER This document was prepared as an account of work sponsored by the United States Government. While this document is believed to contain correct information, neither the United .States Government nor any agency thereof, nor The Regents of the University of California, nor any of their employees, makes any warranty, express or implied, or assumes any legal responsibility for the accuracy, completeness, or usefulness of any information, apparatus, product, or process disclosed, or represents that its use would not infringe privately ()wned rights. Reference he~ein to any specific commercial product, process, or service by its trade name, trademark, manufacturer, or otherwise, does not necessarily constitute or imply its endorsement, recommendation, or favoring by the United States Government or any agency thereof, or The Regents of the University of California. The views and opinions of authors expressed herein do not necessarily state or reflect those of the United States Government or any agency thereof, or The Regents of the University of California. -

Chapter 7 Periodic Properties of the Elements Learning Outcomes
Chapter 7 Periodic Properties of the Elements Learning Outcomes: Explain the meaning of effective nuclear charge, Zeff, and how Zeff depends on nuclear charge and electron configuration. Predict the trends in atomic radii, ionic radii, ionization energy, and electron affinity by using the periodic table. Explain how the radius of an atom changes upon losing electrons to form a cation or gaining electrons to form an anion. Write the electron configurations of ions. Explain how the ionization energy changes as we remove successive electrons, and the jump in ionization energy that occurs when the ionization corresponds to removing a core electron. Explain how irregularities in the periodic trends for electron affinity can be related to electron configuration. Explain the differences in chemical and physical properties of metals and nonmetals, including the basicity of metal oxides and the acidity of nonmetal oxides. Correlate atomic properties, such as ionization energy, with electron configuration, and explain how these relate to the chemical reactivity and physical properties of the alkali and alkaline earth metals (groups 1A and 2A). Write balanced equations for the reactions of the group 1A and 2A metals with water, oxygen, hydrogen, and the halogens. List and explain the unique characteristics of hydrogen. Correlate the atomic properties (such as ionization energy, electron configuration, and electron affinity) of group 6A, 7A, and 8A elements with their chemical reactivity and physical properties. Development of Periodic Table •Dmitri Mendeleev and Lothar Meyer (~1869) independently came to the same conclusion about how elements should be grouped in the periodic table. •Henry Moseley (1913) developed the concept of atomic numbers (the number of protons in the nucleus of an atom) 1 Predictions and the Periodic Table Mendeleev, for instance, predicted the discovery of germanium (which he called eka-silicon) as an element with an atomic weight between that of zinc and arsenic, but with chemical properties similar to those of silicon. -

Quantum-Mechanical Relation Between Atomic Dipole Polarizability and the Van Der Waals Radius (Supplemental Material)
Quantum-Mechanical Relation between Atomic Dipole Polarizability and the van der Waals Radius Dmitry V. Fedorov,1, ∗ Mainak Sadhukhan,1 Martin St¨ohr,1 and Alexandre Tkatchenko1 1Physics and Materials Science Research Unit, University of Luxembourg, L-1511 Luxembourg The atomic dipole polarizability, α, and the van der Waals (vdW) radius, RvdW, are two key quantities to describe vdW interactions between atoms in molecules and materials. Until now, they have been determined independently and separately from each other. Here, we derive the quantum- 1/7 mechanical relation RvdW = const. × α which is markedly different from the common assumption 1/3 RvdW ∝ α based on a classical picture of hard-sphere atoms. As shown for 72 chemical elements between hydrogen and uranium, the obtained formula can be used as a unified definition of the vdW radius solely in terms of the atomic polarizability. For vdW-bonded heteronuclear dimers consisting of atoms A and B, the combination rule α = (αA + αB )/2 provides a remarkably accurate way to calculate their equilibrium interatomic distance. The revealed scaling law allows to reduce the empiricism and improve the accuracy of interatomic vdW potentials, at the same time suggesting the existence of a non-trivial relation between length and volume in quantum systems. The idea to use a specific radius, describing a distance by Bondi [8] has been extensively used. However, it is an atom maintains from other atoms in non-covalent in- based on a restricted amount of experimental informa- teractions, was introduced by Mack [1] and Magat [2]. tion available at that time. With the improvement of Subsequently, it was employed by Kitaigorodskii in his experimental techniques and increase of available data, theory of close packing of molecules in crystals [3, 4]. -

Actinide Overview
2 Meet the Presenter… Alena Paulenova Dr. Alena Paulenova is Associate Professor in the Department of Nuclear Engineering and Director of the Laboratory of Transuranic Elements at the OSU Radiation Center. She is also Adjunct Professor at the Department of Chemistry at Oregon State University , a Joint Research faculty with Idaho National Laboratory, Division of Aqueous Separations and Radiochemistry and a member of the INEST Fuel Cycle Core Committee. She received her Ph.D. in Physical Chemistry in 1985 from the Moscow/Kharkov State University. Until 1999, she was a faculty member at the Department of Nuclear Chemistry and Radioecology of Comenius University in Bratislava, then a visiting scientist at Clemson University and Washington State University in Pullman. In 2003 she joined the faculty at OSU as a Coordinator of the Radiochemistry Program at OSU Radiation Center to bring her experience to the task of helping to educate a new generation of radiochemists: http://oregonstate.edu/~paulenoa/. Her research interest has focused on application of radioanalytical and spectroscopic methods to speciation of radionuclides in aqueous and organic solutions and development of separation methods for spent nuclear fuel cycle processing, decontamination and waste minimization. The main efforts of her research group are fundamental studies of the kinetics and thermodynamics of the complexation of metals, primary actinides and fission products, with organic and inorganic ligands and interactions with redox active species, and the effects of radiolysis and hydrolysis in these systems. Contact: (+1) 541-737-7070 E-mail: [email protected]. An Overview of Actinide Chemistry Alena Paulenova National Analytical Management Program (NAMP) U.S. -

The Ionic Radius of No3+
Journal of Nuclear and Radiochemical Sciences,Vol. 3, No. 1, pp. 147–149, 2002 147 The Ionic Radius of No3+ A. Bilewicz∗ Department of Radiochemistry, Institute of Nuclear Chemistry and Technology, Dorodna 16, 03-195 Warsaw, Poland Received: November 13, 2001; In Final Form: April 30, 2002 259 248 18 5+ Nobelium ( No, T1/2 = 58 min) was produced in bombardment of Cm target with O ions. Next, it was oxi- dised by H5IO6 and loaded together with lanthanide tracers on a chromatographic column filled with cryptomelane 2+ MnO2. In the HNO3 elution curve two peaks are observed. First one is related to the nonoxidized No and the second corresponds to No3+ close to positions of elution peaks of Ho3+ and Y3+. The ionic radius deduced from the elution position of No3+ is to be 89.4 ± 0.7 pm. 1. Introduction to possess interesting ion exchange properties. The cryptome- lane MnO2 phase has a well-defined 2 × 2 tunnel-framed struc- Studies of the heaviest actinides and comparison with the ture with exchangeable hydrogen, alkali, or alkali earth cations.6 properties of lanthanides are interesting because they help to The presence of exchangeable ions in the tunnels requires that understand the influence of relativistic effects on the chemical some of the manganese atoms must be present in oxidation properties. The relativistic effects influence the contraction of states lower than 4. The formulae could be represented by the 3+ lanthanides and actinides ionic radii by two ways: split- IV III 7 HMn7 Mn O16 as postulated by Tsuji and Tamaura. -

Recent Developments in Chalcogen Chemistry
RECENT DEVELOPMENTS IN CHALCOGEN CHEMISTRY Tristram Chivers Department of Chemistry, University of Calgary, Calgary, Alberta, Canada WHERE IS CALGARY? Lecture 1: Background / Introduction Outline • Chalcogens (O, S, Se, Te, Po) • Elemental Forms: Allotropes • Uses • Trends in Atomic Properties • Spin-active Nuclei; NMR Spectra • Halides as Reagents • Cation Formation and Stabilisation • Anions: Structures • Solutions of Chalcogens in Ionic Liquids • Oxides and Imides: Multiple Bonding 3 Elemental Forms: Sulfur Allotropes Sulfur S6 S7 S8 S10 S12 S20 4 Elemental Forms: Selenium and Tellurium Allotropes Selenium • Grey form - thermodynamically stable: helical structure cf. plastic sulfur. R. Keller, et al., Phys. Rev. B. 1977, 4404. • Red form - cyclic Cyclo-Se8 (cyclo-Se7 and -Se6 also known). Tellurium • Silvery-white, metallic lustre; helical structure, cf. grey Se. • Cyclic allotropes only known entrapped in solid-state structures e.g. Ru(Ten)Cl3 (n = 6, 8, 9) M. Ruck, Chem. Eur. J. 2011, 17, 6382 5 Uses – Sulfur Sulfur : Occurs naturally in underground deposits. • Recovered by Frasch process (superheated water). • H2S in sour gas (> 70%): Recovered by Klaus process: Klaus Process: 2 H S + SO 3/8 S + 2 H O 2 2 8 2 • Primary industrial use (70 %): H2SO4 in phosphate fertilizers 6 Uses – Selenium and Tellurium Selenium and Tellurium : Recovered during the refining of copper sulfide ores Selenium: • Photoreceptive properties – used in photocopiers (As2Se3) • Imparts red color in glasses Tellurium: • As an alloy with Cu, Fe, Pb and to harden