Comprehensive Variation Discovery and Recovery of Missing Sequence in the Pig Genome Using Multiple De Novo Assemblies
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Seq2pathway Vignette
seq2pathway Vignette Bin Wang, Xinan Holly Yang, Arjun Kinstlick May 19, 2021 Contents 1 Abstract 1 2 Package Installation 2 3 runseq2pathway 2 4 Two main functions 3 4.1 seq2gene . .3 4.1.1 seq2gene flowchart . .3 4.1.2 runseq2gene inputs/parameters . .5 4.1.3 runseq2gene outputs . .8 4.2 gene2pathway . 10 4.2.1 gene2pathway flowchart . 11 4.2.2 gene2pathway test inputs/parameters . 11 4.2.3 gene2pathway test outputs . 12 5 Examples 13 5.1 ChIP-seq data analysis . 13 5.1.1 Map ChIP-seq enriched peaks to genes using runseq2gene .................... 13 5.1.2 Discover enriched GO terms using gene2pathway_test with gene scores . 15 5.1.3 Discover enriched GO terms using Fisher's Exact test without gene scores . 17 5.1.4 Add description for genes . 20 5.2 RNA-seq data analysis . 20 6 R environment session 23 1 Abstract Seq2pathway is a novel computational tool to analyze functional gene-sets (including signaling pathways) using variable next-generation sequencing data[1]. Integral to this tool are the \seq2gene" and \gene2pathway" components in series that infer a quantitative pathway-level profile for each sample. The seq2gene function assigns phenotype-associated significance of genomic regions to gene-level scores, where the significance could be p-values of SNPs or point mutations, protein-binding affinity, or transcriptional expression level. The seq2gene function has the feasibility to assign non-exon regions to a range of neighboring genes besides the nearest one, thus facilitating the study of functional non-coding elements[2]. Then the gene2pathway summarizes gene-level measurements to pathway-level scores, comparing the quantity of significance for gene members within a pathway with those outside a pathway. -

Análise Integrativa De Perfis Transcricionais De Pacientes Com
UNIVERSIDADE DE SÃO PAULO FACULDADE DE MEDICINA DE RIBEIRÃO PRETO PROGRAMA DE PÓS-GRADUAÇÃO EM GENÉTICA ADRIANE FEIJÓ EVANGELISTA Análise integrativa de perfis transcricionais de pacientes com diabetes mellitus tipo 1, tipo 2 e gestacional, comparando-os com manifestações demográficas, clínicas, laboratoriais, fisiopatológicas e terapêuticas Ribeirão Preto – 2012 ADRIANE FEIJÓ EVANGELISTA Análise integrativa de perfis transcricionais de pacientes com diabetes mellitus tipo 1, tipo 2 e gestacional, comparando-os com manifestações demográficas, clínicas, laboratoriais, fisiopatológicas e terapêuticas Tese apresentada à Faculdade de Medicina de Ribeirão Preto da Universidade de São Paulo para obtenção do título de Doutor em Ciências. Área de Concentração: Genética Orientador: Prof. Dr. Eduardo Antonio Donadi Co-orientador: Prof. Dr. Geraldo A. S. Passos Ribeirão Preto – 2012 AUTORIZO A REPRODUÇÃO E DIVULGAÇÃO TOTAL OU PARCIAL DESTE TRABALHO, POR QUALQUER MEIO CONVENCIONAL OU ELETRÔNICO, PARA FINS DE ESTUDO E PESQUISA, DESDE QUE CITADA A FONTE. FICHA CATALOGRÁFICA Evangelista, Adriane Feijó Análise integrativa de perfis transcricionais de pacientes com diabetes mellitus tipo 1, tipo 2 e gestacional, comparando-os com manifestações demográficas, clínicas, laboratoriais, fisiopatológicas e terapêuticas. Ribeirão Preto, 2012 192p. Tese de Doutorado apresentada à Faculdade de Medicina de Ribeirão Preto da Universidade de São Paulo. Área de Concentração: Genética. Orientador: Donadi, Eduardo Antonio Co-orientador: Passos, Geraldo A. 1. Expressão gênica – microarrays 2. Análise bioinformática por module maps 3. Diabetes mellitus tipo 1 4. Diabetes mellitus tipo 2 5. Diabetes mellitus gestacional FOLHA DE APROVAÇÃO ADRIANE FEIJÓ EVANGELISTA Análise integrativa de perfis transcricionais de pacientes com diabetes mellitus tipo 1, tipo 2 e gestacional, comparando-os com manifestações demográficas, clínicas, laboratoriais, fisiopatológicas e terapêuticas. -

Aneuploidy: Using Genetic Instability to Preserve a Haploid Genome?
Health Science Campus FINAL APPROVAL OF DISSERTATION Doctor of Philosophy in Biomedical Science (Cancer Biology) Aneuploidy: Using genetic instability to preserve a haploid genome? Submitted by: Ramona Ramdath In partial fulfillment of the requirements for the degree of Doctor of Philosophy in Biomedical Science Examination Committee Signature/Date Major Advisor: David Allison, M.D., Ph.D. Academic James Trempe, Ph.D. Advisory Committee: David Giovanucci, Ph.D. Randall Ruch, Ph.D. Ronald Mellgren, Ph.D. Senior Associate Dean College of Graduate Studies Michael S. Bisesi, Ph.D. Date of Defense: April 10, 2009 Aneuploidy: Using genetic instability to preserve a haploid genome? Ramona Ramdath University of Toledo, Health Science Campus 2009 Dedication I dedicate this dissertation to my grandfather who died of lung cancer two years ago, but who always instilled in us the value and importance of education. And to my mom and sister, both of whom have been pillars of support and stimulating conversations. To my sister, Rehanna, especially- I hope this inspires you to achieve all that you want to in life, academically and otherwise. ii Acknowledgements As we go through these academic journeys, there are so many along the way that make an impact not only on our work, but on our lives as well, and I would like to say a heartfelt thank you to all of those people: My Committee members- Dr. James Trempe, Dr. David Giovanucchi, Dr. Ronald Mellgren and Dr. Randall Ruch for their guidance, suggestions, support and confidence in me. My major advisor- Dr. David Allison, for his constructive criticism and positive reinforcement. -

TBCA (1-108) Human Protein – AR09214PU-N | Origene
OriGene Technologies, Inc. 9620 Medical Center Drive, Ste 200 Rockville, MD 20850, US Phone: +1-888-267-4436 [email protected] EU: [email protected] CN: [email protected] Product datasheet for AR09214PU-N TBCA (1-108) Human Protein Product data: Product Type: Recombinant Proteins Description: TBCA (1-108) human recombinant protein, 0.1 mg Species: Human Expression Host: E. coli Predicted MW: 12.8 kDa Concentration: lot specific Purity: >95% by SDS - PAGE Buffer: Presentation State: Purified State: Liquid purified protein Buffer System: 20 mM Tris-HCl buffer pH 7.5 containing 1 mM DTT, 10% glycerol Preparation: Liquid purified protein Protein Description: Recombinant human TBCA protein was expressed in E.coli and purified by using conventional chromatography techniques. Storage: Store undiluted at 2-8°C for up to two weeks or (in aliquots) at -20°C or -70°C for longer. Avoid repeated freezing and thawing. Stability: Shelf life: one year from despatch. RefSeq: NP_001284667 Locus ID: 6902 UniProt ID: O75347 Cytogenetics: 5q14.1 Summary: The product of this gene is one of four proteins (cofactors A, D, E, and C) involved in the pathway leading to correctly folded beta-tubulin from folding intermediates. Cofactors A and D are believed to play a role in capturing and stabilizing beta-tubulin intermediates in a quasi-native confirmation. Cofactor E binds to the cofactor D/beta-tubulin complex; interaction with cofactor C then causes the release of beta-tubulin polypeptides that are committed to the native state. This gene encodes chaperonin cofactor A. Multiple alternatively spliced transcript variants encoding different isoforms have been found for this gene. -

Identification of Potential Pathogenic Genes Associated with Osteoporosis
610.BJBJR0010.1302/2046-3758.612.BJR-2017-0102 research-article2017 Freely available online OPEN ACCESS BJR RESEARCH Identification of potential pathogenic genes associated with osteoporosis Objectives B. Xia, Osteoporosis is a chronic disease. The aim of this study was to identify key genes in osteo- Y. Li, porosis. J. Zhou, Methods B. Tian, Microarray data sets GSE56815 and GSE56814, comprising 67 osteoporosis blood samples L. Feng and 62 control blood samples, were obtained from the Gene Expression Omnibus database. Differentially expressed genes (DEGs) were identified in osteoporosis using Limma pack- Jining No. 1 People’s age (3.2.1) and Meta-MA packages. Gene Ontology and Kyoto Encyclopedia of Genes and Hospital, Jining, Genomes enrichment analyses were performed to identify biological functions. Further- Shandong Province, more, the transcriptional regulatory network was established between the top 20 DEGs and China transcriptional factors using the UCSC ENCODE Genome Browser. Receiver operating char- acteristic (ROC) analysis was applied to investigate the diagnostic value of several DEGs. Results A total of 1320 DEGs were obtained, of which 855 were up-regulated and 465 were down- regulated. These differentially expressed genes were enriched in Gene Ontology terms and Kyoto Encyclopedia of Genes and Genomes pathways, mainly associated with gene expres- sion and osteoclast differentiation. In the transcriptional regulatory network, there were 6038 interactions pairs involving 88 transcriptional factors. In addition, the quantitative reverse transcriptase-polymerase chain reaction result validated the expression of several genes (VPS35, FCGR2A, TBCA, HIRA, TYROBP, and JUND). Finally, ROC analyses showed that VPS35, HIRA, PHF20 and NFKB2 had a significant diagnostic value for osteoporosis. -
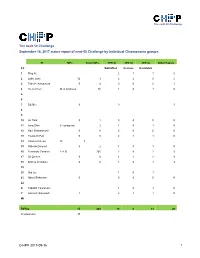
Next-MP50 Status Report
The neXt-50 Challenge The neXt-50 Challenge September 16, 2017 status report of next-50 Challenge by individual Chromosome groups. PI MPs Silver MPs JPR SI JPR SI JPR SI Other Papers Ch Submitted In press In revision 1 Ping Xu 2 1 1 0 2 Lydie Lane 12 3 2 2 0 2 3 Takeshi Kawamura 0 4 0 0 0 1 4 Yu-Ju Chen 26 in progress 86 1 0 1 0 5 6 7 Ed Nice 0 0 1 8 9 10 Jin Park 0 1 0 0 0 0 11 Jong Shin 2 in progress 2 1 0 1 0 12 Ravi Sirdeshmukh 0 0 0 0 0 0 13 Young-Ki Paik 0 5 2 1 1 0 14 CharLes Pineau 12 3 15 GiLberto Domont 0 2 1 0 1 0 16 Fernando CorraLes 2 (+ 9) 165 2 0 2 5 17 GiL Omenn 0 0 3 1 2 8 18 Andrey Archakov 0 0 1 0 1 3 19 20 Siqi Liu 1 0 1 21 Albert Sickmann 0 0 0 0 0 22 X Tadashi Yamamoto 1 0 1 0 Y Hosseini SaLekdeh 1 2 1 1 0 Mt TOTAL 15 268 19 6 13 20 (in progress) 37 C-HPP 2017-09-16 1 The neXt-50 Challenge Chromosome 1 (Ping Xu) PIC Leaders: Ping Xu, Fuchu He Contributing labs: Ping Xu, Beijing Proteome Research Center Fuchu He, Beijing Proteome Research Center Dong Yang, Beijing Proteome Research Center Wantao Ying, Beijing Proteome Research Center Pengyuan Yang, Fudan University Siqi Liu, Beijing Genome Institute Qinyu He, Jinan University Major lab members or partners contributing to the neXt50: Yao Zhang (Beijing Proteome Research Center), Yihao Wang (Beijing Proteome Research Center), Cuitong He (Beijing Proteome Research Center), Wei Wei (Beijing Proteome Research Center), Yanchang Li (Beijing Proteome Research Center), Feng Xu (Beijing Proteome Research Center), Xuehui Peng (Beijing Proteome Research Center). -

Supplemental Figure and Table Legends
Supplemental figure and table legends Supplementary Figure 1: KIAA1841 is well conserved among vertebrates. NCBI HomoloGene pairwise alignment scores of human KIAA1841 sequence compared to other vertebrate orthologs. Supplementary Figure 2: µ-germline transcripts (GLT) and AID mRNA expression are not affected by overexpression of KIAA1841. Splenic B cells were isolated from wild-type mice, and transduced with retroviral vector control (pMIG) or a vector expressing KIAA1841. Levels of µ-GLT and AID mRNA were determined at 72h post-infection by RT-qPCR, and normalized to -actin mRNA and the pMIG control. The mean of three independent experiments +/- SD is shown. NS, p = not significant, p 0.05, two-tailed paired student’s t-test. Supplementary Figure 3: Overexpression of untagged and Xpress-tagged KIAA1841 does not affect cell proliferation. Splenic B cells were isolated from wild-type mice, stimulated with LPS+IL4, and transduced with retroviral vector control (pMIG) or a vector expressing KIAA1841 or Xpress (Xp)-tagged KIAA1841. Cells are labeled with seminaphthorhodafluor (SNARF) cell tracking dye and SNARF intensity was measured at 0h, 24h, and 48h after retroviral infection. Histograms of transduced cells (GFP+) for pMIG control, KIAA1841 and Xp-KIAA1841 were superimposed at each time point. Three independent retroviral infection experiments are shown. Supplementary Figure 4: Sequence alignment of the putative SANT domain of KIAA1841 with the SANT domain of SWI3. Alignment was performed using ClustalOmega; *, conserved residue, :, strongly similar residues, ., weakly similar residues. Numbers indicate amino acid residues in each sequence. Helix 3, which has been reported to be important for the chromatin remodeling function of SWI3 (Boyer et. -

Homotypic Clusters of Transcription Factor Binding Sites Are a Key Component of Human Promoters and Enhancers
Downloaded from genome.cshlp.org on October 4, 2021 - Published by Cold Spring Harbor Laboratory Press Research Homotypic clusters of transcription factor binding sites are a key component of human promoters and enhancers Valer Gotea,1 Axel Visel,2,3 John M. Westlund,4 Marcelo A. Nobrega,4 Len A. Pennacchio,2,3 and Ivan Ovcharenko1,5 1National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, Maryland 20894, USA; 2Genomics Division, Lawrence Berkeley National Laboratory, Berkeley, California 94720, USA; 3United States Department of Energy Joint Genome Institute, Walnut Creek, California 94598, USA; 4Department of Human Genetics, University of Chicago, Chicago, Illinois 60637, USA Clustering of multiple transcription factor binding sites (TFBSs) for the same transcription factor (TF) is a common feature of cis-regulatory modules in invertebrate animals, but the occurrence of such homotypic clusters of TFBSs (HCTs) in the human genome has remained largely unknown. To explore whether HCTs are also common in human and other verte- brates, we used known binding motifs for vertebrate TFs and a hidden Markov model–based approach to detect HCTs in the human, mouse, chicken, and fugu genomes, and examined their association with cis-regulatory modules. We found that evolutionarily conserved HCTs occupy nearly 2% of the human genome, with experimental evidence for individual TFs supporting their binding to predicted HCTs. More than half of the promoters of human genes contain HCTs, with a dis- tribution around the transcription start site in agreement with the experimental data from the ENCODE project. In addition, almost half of the 487 experimentally validated developmental enhancers contain them as well—a number more than 25-fold larger than expected by chance. -

Identification of Gene Biomarkers for Distinguishing Small-Cell Lung Cancer from Non-Small-Cell Lung Cancer Using a Network-Based Approach
Hindawi Publishing Corporation BioMed Research International Volume 2015, Article ID 685303, 8 pages http://dx.doi.org/10.1155/2015/685303 Research Article Identification of Gene Biomarkers for Distinguishing Small-Cell Lung Cancer from Non-Small-Cell Lung Cancer Using a Network-Based Approach Fei Long,1 Jia-Hang Su,2 Bin Liang,1 Li-Li Su,1 and Shu-Juan Jiang1 1 Department of Respiratory Medicine, Shandong Provincial Hospital Affiliated to Shandong University, Jinan, Shandong 250021, China 2Department of Pharmacy, Yantai Chinese Medicine Hospital, Yantai, Shandong 264100, China Correspondence should be addressed to Li-Li Su; [email protected] and Shu-Juan Jiang; [email protected] Received 12 January 2015; Revised 10 March 2015; Accepted 17 March 2015 AcademicEditor:XiaoChang Copyright © 2015 Fei Long et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Lung cancer consists of two main subtypes: small-cell lung cancer (SCLC) and non-small-cell lung cancer (NSCLC) that are classified according to their physiological phenotypes. In this study, we have developed a network-based approach to identify molecular biomarkers that can distinguish SCLC from NSCLC. By identifying positive and negative coexpression gene pairs in normal lung tissues, SCLC, or NSCLC samples and using functional association information from the STRING network, we first construct a lung cancer-specific gene association network. From the network, we obtain gene modules in which genesare highly functionally associated with each other and are either positively or negatively coexpressed in the three conditions. -

Quinalizarin As a Potent, Selective and Cell Permeable Inhibitor of Protein
Quinalizarin as a potent, selective and cell permeable inhibitor of protein kinase CK2 Giorgio Cozza, Marco Mazzorana, Elena Papinutto, Jenny Bain, Matthew Elliott, Giovanni Di Maira, Alessandra Gianoncelli, Mario A. Pagano, Stefania Sarno, Maria Ruzzene, et al. To cite this version: Giorgio Cozza, Marco Mazzorana, Elena Papinutto, Jenny Bain, Matthew Elliott, et al.. Quinalizarin as a potent, selective and cell permeable inhibitor of protein kinase CK2. Biochemical Journal, Port- land Press, 2009, 421 (3), pp.387-395. 10.1042/BJ20090069. hal-00479150 HAL Id: hal-00479150 https://hal.archives-ouvertes.fr/hal-00479150 Submitted on 30 Apr 2010 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Biochemical Journal Immediate Publication. Published on 11 May 2009 as manuscript BJ20090069 QUINALIZARIN AS A POTENT, SELECTIVE AND CELL PERMEABLE INHIBITOR OF PROTEIN KINASE CK2. Giorgio COZZA *, Marco MAZZORANA *† , Elena PAPINUTTO †, Jenny BAIN §, Matthew ELLIOTT §, Giovanni DI MAIRA *† , Alessandra GIANONCELLI *, Mario A. PAGANO *† , Stefania SARNO *† , Maria -

Mouse Anti Human Tubulin Folding Cofactor A(Clone:PAT1A5AT.)
ABGENEX Pvt. Ltd., E-5, Infocity, KIIT Post Office, Tel : +91-674-2720712, +91-9437550560 Email : [email protected] Bhubaneswar, Odisha - 751024, INDIA 32-6228: Mouse Anti Human Tubulin Folding Cofactor A(Clone:PAT1A5AT.) Clonality : Monoclonal Clone Name : PAT1A5AT. Application : ELISA,WB Gene : TERF2IP Gene ID : 54386 Uniprot ID : Q9NYB0 Format : Purified Alternative Name : Tubulin-specific chaperone A,Tubulin-folding cofactor A,CFA,TCP1-chaperonin cofactor A,TBCA. Isotype : Mouse IgG2b heavy chain and ? light chain. Immunogen Information : Anti-human TBCA mAb is derived from hybridization of mouse F0 myeloma cells with spleen cells from BALB/c mice immunized with recombinant human TBCA amino acids 1-108 purified from E. coli. Description TBCA is a tubulin-folding protein which is involved in the early step of the tubulin folding pathway. TBCA is one of four proteins (cofactors A, D, E, and C) implicated in the pathway directing to properly folded beta-tubulin from folding intermediates. Cofactors A and D are thought to be a factor in capturing and stabilizing beta-tubulin in a quasi-native confirmation. TBCA is crucial for cell viability, if reduced it causes a decrease in the amount of soluble tubulin, alterations in microtubules and G1 cell cycle arrest. Cofactor E attaches to the cofactor D-tubulin complex, afterward, interaction with cofactor C triggers the release of tubulin polypeptides that are committed to the native state. Product Info Amount : 20 µg Purification : TBCA antibody was purified from mouse ascitic fluids by protein-G affinity chromatography. Content : 1mg/ml containing PBS, pH-7.4, & 0.1% Sodium Azide. -

Phylogeny, Transposable Element and Sex Chromosome Evolution of The
bioRxiv preprint doi: https://doi.org/10.1101/750109; this version posted August 28, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Phylogeny, transposable element and sex chromosome evolution of the basal lineage of birds Zongji Wang1,2,3,* Jilin Zhang4,*, Xiaoman Xu1, Christopher Witt5, Yuan Deng3, Guangji Chen3, Guanliang Meng3, Shaohong Feng3, Tamas Szekely6,7, Guojie Zhang3,8,9,10,#, Qi Zhou1,2,11,# 1. MOE Laboratory of Biosystems Homeostasis & Protection, Life Sciences Institute, Zhejiang University, Hangzhou, 310058, China 2. Department of Molecular Evolution and Development, University of Vienna, Vienna, 1090, Austria 3. BGI-Shenzhen, Beishan Industrial Zone, Shenzhen 518083, China 4. Department of Medical Biochemistry and Biophysics, Karolinska Institutet, SE-171 77, Stockholm, Sweden 5. Department of Biology and the Museum of Southwestern Biology, University of New Mexico, Albuquerque, NM 87131, USA 6. State Key Laboratory of Biocontrol, Department of Ecology, School of Life Sciences, Sun Yat-sen University, Guangzhou, 510275, China 7. Milner Center for Evolution, Department of Biology and Biochemistry, University of Bath, Bath, BA1 7AY, UK 8 State Key Laboratory of Genetic Resources and Evolution, Kunming Institute of Zoology, Chinese Academy of Sciences, Kunming 650223, China 9. Section for Ecology and Evolution, Department of Biology, University of Copenhagen, DK-2100 Copenhagen, Denmark 10. Center for Excellence in Animal Evolution and Genetics, Chinese Academy of Sciences, Kunming, 650223, China 11. Center for Reproductive Medicine, The 2nd Affiliated Hospital, School of Medicine, Zhejiang University * These authors contributed equally to the work.