Imaging and Single Cell Sequencing Analyses of Super-Enhancer Activation Mediated by NF-Κb in B Cells
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Effects of Rapamycin on Social Interaction Deficits and Gene
Kotajima-Murakami et al. Molecular Brain (2019) 12:3 https://doi.org/10.1186/s13041-018-0423-2 RESEARCH Open Access Effects of rapamycin on social interaction deficits and gene expression in mice exposed to valproic acid in utero Hiroko Kotajima-Murakami1,2, Toshiyuki Kobayashi3, Hirofumi Kashii1,4, Atsushi Sato1,5, Yoko Hagino1, Miho Tanaka1,6, Yasumasa Nishito7, Yukio Takamatsu7, Shigeo Uchino1,2 and Kazutaka Ikeda1* Abstract The mammalian target of rapamycin (mTOR) signaling pathway plays a crucial role in cell metabolism, growth, and proliferation. The overactivation of mTOR has been implicated in the pathogenesis of syndromic autism spectrum disorder (ASD), such as tuberous sclerosis complex (TSC). Treatment with the mTOR inhibitor rapamycin improved social interaction deficits in mouse models of TSC. Prenatal exposure to valproic acid (VPA) increases the incidence of ASD. Rodent pups that are exposed to VPA in utero have been used as an animal model of ASD. Activation of the mTOR signaling pathway was recently observed in rodents that were exposed to VPA in utero, and rapamycin ameliorated social interaction deficits. The present study investigated the effect of rapamycin on social interaction deficits in both adolescence and adulthood, and gene expressions in mice that were exposed to VPA in utero. We subcutaneously injected 600 mg/kg VPA in pregnant mice on gestational day 12.5 and used the pups as a model of ASD. The pups were intraperitoneally injected with rapamycin or an equal volume of vehicle once daily for 2 consecutive days. The social interaction test was conducted in the offspring after the last rapamycin administration at 5–6 weeks of ages (adolescence) or 10–11 weeks of age (adulthood). -

Ribosomal RNA‑Depleted RNA Sequencing Reveals the Pathogenesis of Refractory Mycoplasma Pneumoniae Pneumonia in Children
MOLECULAR MEDICINE REPORTS 24: 761, 2021 Ribosomal RNA‑depleted RNA sequencing reveals the pathogenesis of refractory Mycoplasma pneumoniae pneumonia in children FENG HUANG1,2*, HUIFENG FAN1*, DIYUAN YANG1, JUNSONG ZHANG3, TINGTING SHI1, DONGWEI ZHANG1 and GEN LU1 1Department of Respiration, Guangzhou Women and Children's Medical Center, Guangzhou Medical University, Guangzhou, Guangdong 510120; 2Institute of Human Virology, Zhongshan School of Medicine, Sun Yat‑sen University; 3Guangdong Provincial People's Hospital, Guangdong Academy of Medical Sciences, Guangzhou, Guangdong 510080, P.R. China Received April 17, 2020; Accepted May 28, 2021 DOI: 10.3892/mmr.2021.12401 Abstract. Pneumonia caused by Mycoplasma pneumoniae in the NRMPP and RMPP comparative groups were primarily (M. pneumoniae) is a major cause of community‑acquired enriched in ‘herpes simplex virus 1 infection’, ‘viral carcinogen‑ pneumonia in children. In some cases, M. pneumoniae pneu‑ esis’ and ‘RNA transport’. In the present study, a comprehensive monia (MPP) can develop into refractory MPP (RMPP), which analysis of the differences between the NRMPP and RMPP shows no clinical or radiological response to macrolides, and cases was performed based on rRNA‑depleted RNA‑sequencing can progress to severe and complicated pneumonia. However, techniques, and the selected genes and circRNAs may be closely the pathogenesis of RMPP remains poorly understood. The associated with the complex pathogenesis of RMPP. present study aimed to identify target genes that could be used as biomarkers for the clinical diagnosis of early‑stage RMPP Introduction through high‑throughput sequencing technology. The differences in long non‑coding (lnc)RNAs, mRNAs and circular (circ)RNAs Mycoplasma pneumoniae (M. pneumoniae) is one of the were examined between whole‑blood samples from two patients main pathogens that cause respiratory tract infections in with non‑refractory MPP (NRMPP), two patients with RMPP humans (1,2). -

Mir-7 in Cancer Development
biomedicines Review MiR-7 in Cancer Development Petra Kora´c 1 , Mariastefania Antica 2 and Maja Matuli´c 1,* 1 Department of Biology, Division of Molecular Biology, Faculty of Science, University of Zagreb, Horvatovac 102, 10000 Zagreb, Croatia; [email protected] 2 Division of Molecular Biology, Rudjer Boskovi´cInstitute, Bijeniˇcka54, 10000 Zagreb, Croatia; [email protected] * Correspondence: [email protected] Abstract: MicroRNAs (miRNAs) are short non-coding RNA involved in the regulation of specific mRNA translation. They participate in cellular signaling circuits and can act as oncogenes in tumor development, so-called oncomirs, as well as tumor suppressors. miR-7 is an ancient miRNA involved in the fine-tuning of several signaling pathways, acting mainly as tumor suppressor. Through downregulation of PI3K and MAPK pathways, its dominant role is the suppression of proliferation and survival, stimulation of apoptosis and inhibition of migration. Besides these functions, it has numerous additional roles in the differentiation process of different cell types, protection from stress and chromatin remodulation. One of the most investigated tissues is the brain, where its downregulation is linked with glioblastoma cell proliferation. Its deregulation is found also in other tumor types, such as in liver, lung and pancreas. In some types of lung and oral carcinoma, it can act as oncomir. miR-7 roles in cell fate determination and maintenance of cell homeostasis are still to be discovered, as well as the possibilities of its use as a specific biotherapeutic. Keywords: microRNAs; miR-7; gene expression; tumor suppressor; cancer cell Citation: Kora´c,P.; Antica, M.; Matuli´c,M. -

Whole Exome Sequencing in Families at High Risk for Hodgkin Lymphoma: Identification of a Predisposing Mutation in the KDR Gene
Hodgkin Lymphoma SUPPLEMENTARY APPENDIX Whole exome sequencing in families at high risk for Hodgkin lymphoma: identification of a predisposing mutation in the KDR gene Melissa Rotunno, 1 Mary L. McMaster, 1 Joseph Boland, 2 Sara Bass, 2 Xijun Zhang, 2 Laurie Burdett, 2 Belynda Hicks, 2 Sarangan Ravichandran, 3 Brian T. Luke, 3 Meredith Yeager, 2 Laura Fontaine, 4 Paula L. Hyland, 1 Alisa M. Goldstein, 1 NCI DCEG Cancer Sequencing Working Group, NCI DCEG Cancer Genomics Research Laboratory, Stephen J. Chanock, 5 Neil E. Caporaso, 1 Margaret A. Tucker, 6 and Lynn R. Goldin 1 1Genetic Epidemiology Branch, Division of Cancer Epidemiology and Genetics, National Cancer Institute, NIH, Bethesda, MD; 2Cancer Genomics Research Laboratory, Division of Cancer Epidemiology and Genetics, National Cancer Institute, NIH, Bethesda, MD; 3Ad - vanced Biomedical Computing Center, Leidos Biomedical Research Inc.; Frederick National Laboratory for Cancer Research, Frederick, MD; 4Westat, Inc., Rockville MD; 5Division of Cancer Epidemiology and Genetics, National Cancer Institute, NIH, Bethesda, MD; and 6Human Genetics Program, Division of Cancer Epidemiology and Genetics, National Cancer Institute, NIH, Bethesda, MD, USA ©2016 Ferrata Storti Foundation. This is an open-access paper. doi:10.3324/haematol.2015.135475 Received: August 19, 2015. Accepted: January 7, 2016. Pre-published: June 13, 2016. Correspondence: [email protected] Supplemental Author Information: NCI DCEG Cancer Sequencing Working Group: Mark H. Greene, Allan Hildesheim, Nan Hu, Maria Theresa Landi, Jennifer Loud, Phuong Mai, Lisa Mirabello, Lindsay Morton, Dilys Parry, Anand Pathak, Douglas R. Stewart, Philip R. Taylor, Geoffrey S. Tobias, Xiaohong R. Yang, Guoqin Yu NCI DCEG Cancer Genomics Research Laboratory: Salma Chowdhury, Michael Cullen, Casey Dagnall, Herbert Higson, Amy A. -

Exploring the Relationship Between Gut Microbiota and Major Depressive Disorders
E3S Web of Conferences 271, 03055 (2021) https://doi.org/10.1051/e3sconf/202127103055 ICEPE 2021 Exploring the Relationship between Gut Microbiota and Major Depressive Disorders Catherine Tian1 1Shanghai American School, Shanghai, China Abstract. Major Depressive Disorder (MDD) is a psychiatric disorder accompanied with a high rate of suicide, morbidity and mortality. With the symptom of an increasing or decreasing appetite, there is a possibility that MDD may have certain connections with gut microbiota, the colonies of microbes which reside in the human digestive system. In recent years, more and more studies started to demonstrate the links between MDD and gut microbiota from animal disease models and human metabolism studies. However, this relationship is still largely understudied, but it is very innovative since functional dissection of this relationship would furnish a new train of thought for more effective treatment of MDD. In this study, by using multiple genetic analytic tools including Allen Brain Atlas, genetic function analytical tools, and MicrobiomeAnalyst, I explored the genes that shows both expression in the brain and the digestive system to affirm that there is a connection between gut microbiota and the MDD. My approach finally identified 7 MDD genes likely to be associated with gut microbiota, implicating 3 molecular pathways: (1) Wnt Signaling, (2) citric acid cycle in the aerobic respiration, and (3) extracellular exosome signaling. These findings may shed light on new directions to understand the mechanism of MDD, potentially facilitating the development of probiotics for better psychiatric disorder treatment. 1 Introduction 1.1 Major Depressive Disorder Major Depressive Disorder (MDD) is a mood disorder that will affect the mood, behavior and other physical parts. -

Ras Functional Proximity Proteomics Establishes Mtorc2 As New Direct Ras Effector
www.oncotarget.com Oncotarget, 2019, Vol. 10, (No. 50), pp: 5126-5135 Research Perspective Ras functional proximity proteomics establishes mTORC2 as new direct ras effector Joanna R. Kovalski1,2, Ronald L. Shanderson1,2, and Paul A. Khavari1,2,3 1Program in Epithelial Biology, Stanford University, Stanford, CA 94305, USA 2Program in Cancer Biology, Stanford University, Stanford, CA 94305, USA 3VA Palo Alto Healthcare System, Palo Alto, CA 94304, USA Correspondence to: Paul A. Khavari, email: [email protected] Keywords: Ras; BioID; proteomics; CRISPR; mTORC2 Received: May 20, 2019 Accepted: May 29, 2019 Published: August 27, 2019 Copyright: Kovalski et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License 3.0 (CC BY 3.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. ABSTRACT Although oncogenic mutations in the three major Ras isoforms, KRAS, HRAS and NRAS, are present in nearly a third of human cancers, therapeutic targeting of Ras remains a challenge due to its structure and complex regulation. However, an in-depth examination of the protein interactome of oncogenic Ras may provide new insights into key regulators, effectors and other mediators of its tumorigenic functions. Previous proteomic analyses have been limited by experimental tools that fail to capture the dynamic, transient nature of Ras cellular interactions. Therefore, in a recent study, we integrated proximity-dependent biotin labeling (BioID) proteomics with CRISPR screening of identified proteins to identify Ras proximal proteins required for Ras-dependent cancer cell growth. Oncogenic Ras was proximal to proteins involved in unexpected biological processes, such as vesicular trafficking and solute transport. -

Mapping of Craniofacial Traits in Outbred Mice Identifies Major Developmental Genes Involved in Shape Determination
Mapping of craniofacial traits in outbred mice identifies major developmental genes involved in shape determination Luisa F Pallares1, Peter Carbonetto2,3, Shyam Gopalakrishnan2,4, Clarissa C Parker2,5, Cheryl L Ackert-Bicknell6, Abraham A Palmer2,7, Diethard Tautz1 # 1Max Planck Institute for Evolutionary Biology, Plön, Germany 2University of Chicago, Chicago, Illinois, USA 3AncestryDNA, San Francisco, California, USA 4Museum of Natural History, Copenhagen University, Copenhagen, Denmark 5Middlebury College, Department of Psychology and Program in Neuroscience, Middlebury VT, USA 6Center for Musculoskeletal Research, University of Rochester, Rochester, NY USA 7University of California San Diego, La Jolla, CA, USA # corresponding author: [email protected] short title: craniofacial shape mapping Abstract The vertebrate cranium is a prime example of the high evolvability of complex traits. While evidence of genes and developmental pathways underlying craniofacial shape determination 1 is accumulating, we are still far from understanding how such variation at the genetic level is translated into craniofacial shape variation. Here we used 3D geometric morphometrics to map genes involved in shape determination in a population of outbred mice (Carworth Farms White, or CFW). We defined shape traits via principal component analysis of 3D skull and mandible measurements. We mapped genetic loci associated with shape traits at ~80,000 candidate single nucleotide polymorphisms in ~700 male mice. We found that craniofacial shape and size are highly heritable, polygenic traits. Despite the polygenic nature of the traits, we identified 17 loci that explain variation in skull shape, and 8 loci associated with variation in mandible shape. Together, the associated variants account for 11.4% of skull and 4.4% of mandible shape variation, however, the total additive genetic variance associated with phenotypic variation was estimated in ~45%. -

Profiling Haplotype Specific Cpg and Cph Methylation Within A
www.nature.com/scientificreports OPEN Profling haplotype specifc CpG and CpH methylation within a schizophrenia GWAS locus on chromosome 14 in schizophrenia and healthy subjects Margarita Alfmova*, Nikolay Kondratyev, Arkadiy Golov & Vera Golimbet Interrogating DNA methylation within schizophrenia risk loci holds promise to identify mechanisms by which genes infuence the disease. Based on the hypothesis that allele specifc methylation (ASM) of a single CpG, or perhaps CpH, might mediate or mark the efects of genetic variants on disease risk and phenotypes, we explored haplotype specifc methylation levels of individual cytosines within a genomic region harbouring the BAG5, APOPT1 and KLC1 genes in peripheral blood of schizophrenia patients and healthy controls. Three DNA fragments located in promoter, intronic and intergenic areas were studied by single-molecule real-time bisulfte sequencing enabling the analysis of long reads of DNA with base-pair resolution and the determination of haplotypes directly from sequencing data. Among 1,012 cytosines studied, we did not fnd any site where methylation correlated with the disease or cognitive defcits after correction for multiple testing. At the same time, we determined the methylation profle associated with the schizophrenia risk haplotype within the KLC1 fourth intron and confrmed ASM for cytosines located in the vicinity of rs67899457. These genetically associated DNA methylation variations may be related to the pathophysiological mechanism diferentiating the risk and non-risk haplotypes and merit further investigation. Schizophrenia is a common, highly heritable disorder characterized by positive, negative, and cognitive symp- toms. Large genome-wide association studies (GWAS) of the Psychiatric Genomics Consortium (PGC) have identifed more than 100 genomic regions that are signifcantly associated with schizophrenia1,2. -

The Human Gene Connectome As a Map of Short Cuts for Morbid Allele Discovery
The human gene connectome as a map of short cuts for morbid allele discovery Yuval Itana,1, Shen-Ying Zhanga,b, Guillaume Vogta,b, Avinash Abhyankara, Melina Hermana, Patrick Nitschkec, Dror Friedd, Lluis Quintana-Murcie, Laurent Abela,b, and Jean-Laurent Casanovaa,b,f aSt. Giles Laboratory of Human Genetics of Infectious Diseases, Rockefeller Branch, The Rockefeller University, New York, NY 10065; bLaboratory of Human Genetics of Infectious Diseases, Necker Branch, Paris Descartes University, Institut National de la Santé et de la Recherche Médicale U980, Necker Medical School, 75015 Paris, France; cPlateforme Bioinformatique, Université Paris Descartes, 75116 Paris, France; dDepartment of Computer Science, Ben-Gurion University of the Negev, Beer-Sheva 84105, Israel; eUnit of Human Evolutionary Genetics, Centre National de la Recherche Scientifique, Unité de Recherche Associée 3012, Institut Pasteur, F-75015 Paris, France; and fPediatric Immunology-Hematology Unit, Necker Hospital for Sick Children, 75015 Paris, France Edited* by Bruce Beutler, University of Texas Southwestern Medical Center, Dallas, TX, and approved February 15, 2013 (received for review October 19, 2012) High-throughput genomic data reveal thousands of gene variants to detect a single mutated gene, with the other polymorphic genes per patient, and it is often difficult to determine which of these being of less interest. This goes some way to explaining why, variants underlies disease in a given individual. However, at the despite the abundance of NGS data, the discovery of disease- population level, there may be some degree of phenotypic homo- causing alleles from such data remains somewhat limited. geneity, with alterations of specific physiological pathways under- We developed the human gene connectome (HGC) to over- come this problem. -

Occurrence and Identification of Emeria Species in Broiler Rearing
Tran et al. Journal of Animal Science and Technology (2015) 57:44 DOI 10.1186/s40781-015-0077-x RESEARCH Open Access In silico characterisation, homology modelling and structure-based functional annotation of blunt snout bream (Megalobrama amblycephala) Hsp70 and Hsc70 proteins Ngoc Tuan Tran1,3, Ivan Jakovlić1 and Wei-Min Wang1,2* Abstract Background: Heat shock proteins play an important role in protection from stress stimuli and metabolic insults in almost all organisms. Methods: In this study, computational tools were used to deeply analyse the physicochemical characteristics and, using homology modelling, reliably predict the tertiary structure of the blunt snout bream (Ma-) Hsp70 and Hsc70 proteins. Derived three-dimensional models were then used to predict the function of the proteins. Results: Previously published predictions regarding the protein length, molecular weight, theoretical isoelectric point and total number of positive and negative residues were corroborated. Among the new findings are: the extinction coefficient (33725/33350 and 35090/34840 - Ma-Hsp70/ Ma-Hsc70, respectively), instability index (33.68/35.56 – both stable), aliphatic index (83.44/80.23 – both very stable), half-life estimates (both relatively stable), grand average of hydropathicity (−0.431/-0.473 – both hydrophilic) and amino acid composition (alanine-lysine-glycine/glycine-lysine-aspartic acid were the most abundant, no disulphide bonds, the N-terminal of both proteins was methionine). Homology modelling was performed by SWISS-MODEL program and the proposed model was evaluated as highly reliable based on PROCHECK’s Ramachandran plot, ERRAT, PROVE, Verify 3D, ProQ and ProSA analyses. Conclusions: The research revealed a high structural similarity to Hsp70 and Hsc70 proteins from several taxonomically distant animal species, corroborating a remarkably high level of evolutionary conservation among the members of this protein family. -

The Human Gene Connectome As a Map of Short Cuts for Morbid Allele Discovery
The human gene connectome as a map of short cuts for morbid allele discovery Yuval Itana,1, Shen-Ying Zhanga,b, Guillaume Vogta,b, Avinash Abhyankara, Melina Hermana, Patrick Nitschkec, Dror Friedd, Lluis Quintana-Murcie, Laurent Abela,b, and Jean-Laurent Casanovaa,b,f aSt. Giles Laboratory of Human Genetics of Infectious Diseases, Rockefeller Branch, The Rockefeller University, New York, NY 10065; bLaboratory of Human Genetics of Infectious Diseases, Necker Branch, Paris Descartes University, Institut National de la Santé et de la Recherche Médicale U980, Necker Medical School, 75015 Paris, France; cPlateforme Bioinformatique, Université Paris Descartes, 75116 Paris, France; dDepartment of Computer Science, Ben-Gurion University of the Negev, Beer-Sheva 84105, Israel; eUnit of Human Evolutionary Genetics, Centre National de la Recherche Scientifique, Unité de Recherche Associée 3012, Institut Pasteur, F-75015 Paris, France; and fPediatric Immunology-Hematology Unit, Necker Hospital for Sick Children, 75015 Paris, France Edited* by Bruce Beutler, University of Texas Southwestern Medical Center, Dallas, TX, and approved February 15, 2013 (received for review October 19, 2012) High-throughput genomic data reveal thousands of gene variants to detect a single mutated gene, with the other polymorphic genes per patient, and it is often difficult to determine which of these being of less interest. This goes some way to explaining why, variants underlies disease in a given individual. However, at the despite the abundance of NGS data, the discovery of disease- population level, there may be some degree of phenotypic homo- causing alleles from such data remains somewhat limited. geneity, with alterations of specific physiological pathways under- We developed the human gene connectome (HGC) to over- come this problem. -
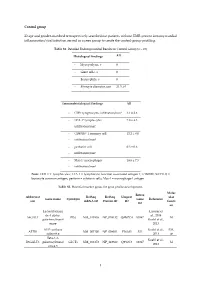
1 Control Group 23 Age and Gender-Matched
Control group 23 age and gender-matched retrospectively searched out patients without EMB-proven intramyocardial inflammation/viral infection served as a peer group to create the control-group profiling. Table S1. Detailed Endomyocardial Results in Control Group (n = 23) Histological findings All - Myocytolysis, n 0 - Giant cells, n 0 - Eosinophils, n 0 - Myocyte diameter, µm 21.3 ± 0 Immunohistological findings All - CD3+ lymphocytes infiltration/mm² 3.1 ± 2.6 - LFA-1+ lymphocytes 7.8 ± 4.3 infiltration/mm² - CD45R0+ T memory cell 12.2 ± 8.0 - infiltration/mm² - perforin+ cell 0.3 ± 0.6 - infiltration/mm² - Mac-1+ macrophages 18.6 ± 7.5 - infiltration/mm² Note: CD3 = T lymphocytes; LFA-1 = lymphocyte function-associated antigen-1; CD45R0 (UCHL1) = leucocyte common antigen; perforin = cytotoxic cells; Mac-1 = macrophage-1 antigen. Table S2. Potential marker genes for gene profile development. Molec Entrez Abbreviat RefSeq RefSeq Uniprot ular Gene name Synonym Gene Reference ion mRNA ID Protein ID ID functi ID on Lactosylcerami Lassner et de 4-alpha- al., 2014 A4GALT P(k) NM_017436 NP_059132 Q9NPC4 53947 M galactosyltransf Kuehl et al., erase 2013 ATP synthase Kuehl et al., EM, ATP5I NM_007100 NP_009031 P56385 521 subunit g 2013 ce Beta-1,3- Kuehl et al., B3GALT5 galactosyltransf GLCT5 NM_033172 NP_149362 Q9Y2C3 10317 M 2013 erase 5 1 BAG family molecular Kuehl et al., BAG5 NM_004873 NP_004864 Q9UL15 9529 CC chaperone 2013 regulator 5 Lassner et CC-chemokine al., 2014 CCL20 ST38 NM_004591 NP_004582 P78556 6364 IR ligand 20 Kuehl et al.,