Principles of Molecular Pathology
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Pseudo-Cholinesterase in Rat Erythrocytes Basis of Its Substrate Ratio, It Is Possible That It May Be a Butyro-Cholinesterase
No. 4535 September 29, 1956 NATURE 697 Pseudo-cholinesterase in Rat Erythrocytes basis of its substrate ratio, it is possible that it may be a butyro-cholinesterase. .ALTHOUGH there are many references in the literature to the existence of different types of cholin D.R. DAVIES esterase in mammalian sera1, it is generally believed J. P. RUTLAND 2 that human red cells contain only one type • Further more, we can find no evidence in the literature for Chemical Defence Experimental Establishment the existence of a pseudo-cholinesterase in any (Ministry of Supply), mammalian erythrocytes, although many workers Porton, Salisbury. have stated that benzoylcholine is hydrolysed by May 3. red cells. Since, however, the rate of hydrolysis is 1 Mundell, D. B., Nature, 153, 557 (1944). Ellis, S., Sanders, S., and slow, this breakdown of a substrate formerly presumed Bodansky, 0., J. Pharmacol., 91, 225 (1947). Levine M G and Suran, A. A., Enzymologia, 15, 17 (1951). ' · ., to be specific for pseudo-cholinesterase has been 'Mendell, B., Mundell, D. B., and Rudney, H., Biochem. J. 37 473 attributed to true cholinesterase. Indeed, Adams3 (1943). ' ' has shown that purified true cholinesterase does • Adams, D. H., Bwchim. Biophys. Acta, ~. 1 (1949). hydrolyse benzoylcholine, at a rate which would • Fraser, P. ~-, ti:,esis, University of Birmingham (1951), and private commumcat10n (1956). account for such observations. In spite of this, • Aldridge, W. N., Biochem. J., 53, 62 (1953). Austin, L., and Berry some results obtained by Fraser with benzoylcholine• W. K., Bwchem. J., 54, 695 (1953). ' imply the existence of more than one type of cholin 'Bayliss, B. -

Aldrich FT-IR Collection Edition I Library
Aldrich FT-IR Collection Edition I Library Library Listing – 10,505 spectra This library is the original FT-IR spectral collection from Aldrich. It includes a wide variety of pure chemical compounds found in the Aldrich Handbook of Fine Chemicals. The Aldrich Collection of FT-IR Spectra Edition I library contains spectra of 10,505 pure compounds and is a subset of the Aldrich Collection of FT-IR Spectra Edition II library. All spectra were acquired by Sigma-Aldrich Co. and were processed by Thermo Fisher Scientific. Eight smaller Aldrich Material Specific Sub-Libraries are also available. Aldrich FT-IR Collection Edition I Index Compound Name Index Compound Name 3515 ((1R)-(ENDO,ANTI))-(+)-3- 928 (+)-LIMONENE OXIDE, 97%, BROMOCAMPHOR-8- SULFONIC MIXTURE OF CIS AND TRANS ACID, AMMONIUM SALT 209 (+)-LONGIFOLENE, 98+% 1708 ((1R)-ENDO)-(+)-3- 2283 (+)-MURAMIC ACID HYDRATE, BROMOCAMPHOR, 98% 98% 3516 ((1S)-(ENDO,ANTI))-(-)-3- 2966 (+)-N,N'- BROMOCAMPHOR-8- SULFONIC DIALLYLTARTARDIAMIDE, 99+% ACID, AMMONIUM SALT 2976 (+)-N-ACETYLMURAMIC ACID, 644 ((1S)-ENDO)-(-)-BORNEOL, 99% 97% 9587 (+)-11ALPHA-HYDROXY-17ALPHA- 965 (+)-NOE-LACTOL DIMER, 99+% METHYLTESTOSTERONE 5127 (+)-P-BROMOTETRAMISOLE 9590 (+)-11ALPHA- OXALATE, 99% HYDROXYPROGESTERONE, 95% 661 (+)-P-MENTH-1-EN-9-OL, 97%, 9588 (+)-17-METHYLTESTOSTERONE, MIXTURE OF ISOMERS 99% 730 (+)-PERSEITOL 8681 (+)-2'-DEOXYURIDINE, 99+% 7913 (+)-PILOCARPINE 7591 (+)-2,3-O-ISOPROPYLIDENE-2,3- HYDROCHLORIDE, 99% DIHYDROXY- 1,4- 5844 (+)-RUTIN HYDRATE, 95% BIS(DIPHENYLPHOSPHINO)BUT 9571 (+)-STIGMASTANOL -

4 Supplementary File
Supplemental Material for High-throughput screening discovers anti-fibrotic properties of Haloperidol by hindering myofibroblast activation Michael Rehman1, Simone Vodret1, Luca Braga2, Corrado Guarnaccia3, Fulvio Celsi4, Giulia Rossetti5, Valentina Martinelli2, Tiziana Battini1, Carlin Long2, Kristina Vukusic1, Tea Kocijan1, Chiara Collesi2,6, Nadja Ring1, Natasa Skoko3, Mauro Giacca2,6, Giannino Del Sal7,8, Marco Confalonieri6, Marcello Raspa9, Alessandro Marcello10, Michael P. Myers11, Sergio Crovella3, Paolo Carloni5, Serena Zacchigna1,6 1Cardiovascular Biology, 2Molecular Medicine, 3Biotechnology Development, 10Molecular Virology, and 11Protein Networks Laboratories, International Centre for Genetic Engineering and Biotechnology (ICGEB), Padriciano, 34149, Trieste, Italy 4Institute for Maternal and Child Health, IRCCS "Burlo Garofolo", Trieste, Italy 5Computational Biomedicine Section, Institute of Advanced Simulation IAS-5 and Institute of Neuroscience and Medicine INM-9, Forschungszentrum Jülich GmbH, 52425, Jülich, Germany 6Department of Medical, Surgical and Health Sciences, University of Trieste, 34149 Trieste, Italy 7National Laboratory CIB, Area Science Park Padriciano, Trieste, 34149, Italy 8Department of Life Sciences, University of Trieste, Trieste, 34127, Italy 9Consiglio Nazionale delle Ricerche (IBCN), CNR-Campus International Development (EMMA- INFRAFRONTIER-IMPC), Rome, Italy This PDF file includes: Supplementary Methods Supplementary References Supplementary Figures with legends 1 – 18 Supplementary Tables with legends 1 – 5 Supplementary Movie legends 1, 2 Supplementary Methods Cell culture Primary murine fibroblasts were isolated from skin, lung, kidney and hearts of adult CD1, C57BL/6 or aSMA-RFP/COLL-EGFP mice (1) by mechanical and enzymatic tissue digestion. Briefly, tissue was chopped in small chunks that were digested using a mixture of enzymes (Miltenyi Biotec, 130- 098-305) for 1 hour at 37°C with mechanical dissociation followed by filtration through a 70 µm cell strainer and centrifugation. -

1-Pyrene-Butyrylcholine: a Fluorescent Probe for the Cholinergic
Proc. Nat. Acad. Sci. USA Vol. 72, No. 8, pp. 3097-3101, August 1975 Biophysics 1-Pyrene-butyrylcholine: A fluorescent probe for the cholinergic system (long-lifetime fluorescent probe/neuromuscular junction/acetylcholine receptor/visualization of synapse) F. J. BARRANTES, B. SAKMANN, R. BONNER, H. EIBL, AND T. M. JOVIN Departments of Molecular Biology, Neurobiology, and Biochemical Kinetics, Max-Planck-Institut fur Biophysikalische Chemie, D-3400 Gottingen-Nikolausberg, West Germany Communicated by Manfred Eigen, May 19,1975 ABSTRACT The action of I-pyrene-butyrylcholine, a probes having intrinsically long excited-state lifetimes along new cholinergic fluorescent probe, as been studied at the with a high sensitivity to environmental factors. With this in cellular level using electrophysiological and fluorescence mind, 1-pyrene-butyrylcholine (PBC) was synthesized, and techniques. The spectroscopic properties of the probe were its action studied at the cellular level under physiological found to be similar to those of pyrene-butyric acid, the ex- cited-state lifetime in air-saturated aqueous solutions being conditions. 92 nsec. At micromolar concentrations the probe was found The fluorescent probe behaves as a nondepolarizing, re- to exert a nondepolarizing, reversible blocking action at the versible blocking agent in a nicotinic synapse, it specifically neuromuscular junction of the frog. The same cholinolytic ef- labels the neuromuscular junction, and it possesses a long ex- fect was observed in hypersensitive denervated muscles. cited-state lifetime. The curare-like cholinergic activity of The synaptic localization of the probe could be observed make it therefore ap- with fluorescence microscopy using sub- and micromolar PBC and its spectroscopic properties concentrations. -

( 12 ) United States Patent
US010131766B2 (12 ) United States Patent (10 ) Patent No. : US 10 , 131, 766 B2 Hazen et al. (45 ) Date of Patent: Nov . 20 , 2018 ( 54 ) UNSATURATED POLYESTER RESIN 6 ,619 ,886 B1 9 /2003 Harrington 6 ,692 , 802 B1 2 / 2004 Nava SYSTEM FOR CURED IN - PLACE PIPING 7 , 135 ,087 B2 11/ 2006 Blackmore et al. 7 ,799 ,228 B2 9 /2010 Bomak et al . (71 ) Applicant : Interplastic Corporation , Saint Paul, 8 , 047, 238 B2 11/ 2011 Wiessner et al . MN (US ) 8 ,053 ,031 B2 11/ 2011 Stanley et al. 8 ,092 ,689 B2 1 / 2012 Gosselin (72 ) Inventors : Benjamin R . Hazen , Roseville , MN 8 , 298 , 360 B2 10 / 2012 Da Silveira et al. 8 ,418 , 728 B1 4 / 2013 Kiest , Jr . (US ) ; David J . Herzog , Maple Grove , 8 ,586 ,653 B2 11 / 2013 Klopsch et al. MN (US ) ; Louis R . Ross, Cincinnati , 8 ,636 , 869 B2 1 / 2014 Wiessner et al . OH (US ) ; Joel R . Weber , Moundsview , 8 ,741 , 988 B2 6 /2014 Klopsch et al. MN (US ) 8 , 877 , 837 B2 11/ 2014 Yu et al. 9 ,068 , 045 B2 6 /2015 Nava et al. 9 ,074 , 040 B2 7 / 2015 Turshani et al. ( 73 ) Assignee : Interplastic Corporation , St. Paul , MN 9 , 150 , 709 B2 10 / 2015 Klopsch et al . (US ) 9 , 207 , 155 B1 12 / 2015 Allouche et al. 9 ,273 ,815 B2 3 / 2016 Gillanders et al. ( * ) Notice : Subject to any disclaimer, the term of this 9 , 371, 950 B2 6 / 2016 Hairston et al. patent is extended or adjusted under 35 9 , 550 , 933 B2 1 /2017 Chatterji et al . -

Butyrylcholinesterase (B7061)
Butyrylcholinesterase human, recombinant expressed in transgenic goats Catalog Number B7061 Storage Temperature –70 °C CAS RN 9001-08-5 Substrates: EC 3.1.1.8 Butyrylthiocholine5 Synonyms: rBChE; Acylcholine acylhydrolase; Butyrylcholine6 Pseudocholinesterase; cholinesterase Acetylcholine7 Acetylthiocholine8 Product Description Cocaine9 Butyrylcholinesterase (BChE) belongs to the same structural class of proteins, the esterase/lipase family, Selective inhibition of BChE activity can be used in the as acetylcholinesterase (AchE, EC 3.1.1.7). They are detection of organophosphates.10 Its use in the serine hydrolases that share substantial structural treatment of organophosphate toxicity shows promise similarities, but differ in substrate specificities and and there is a correlation between the level of BChE in inhibitor sensitivities.1 BChE can, unlike AChE, human blood and degree of protection against efficiently hydrolyze larger esters of choline such as potentially toxic nerve agents.3 butyrylcholine and benzoylcholine. There has also been interest in the roles of BChE cholinesterases with regard to Alzheimer’s disease. Butyrylcholine ® butyric acid + choline Investigations into selective inhibitors may provide a H2O clearer picture of the physiological role of BChE in both healthy and diseased individuals.2 Although BChE is found in the serum, hemopoietic cells, liver, lung, heart, and the central nervous system This recombinant, human product is isolated from the of vertebrates, it has no known physiological function.2,3 milk of transgenic goats. It is purified by affinity chromatography and is primarily in the dimeric form, Molecular mass:4 170 kDa (dimer) with the remainder being monomers. The product is supplied as a solution in 10 mM sodium phosphate, Extinction coefficient:4 E1% = 19.8 (280 nm) pH 7.2, with 50 mM NaCl, 1 mM EDTA, and 10% glycerol. -

The Role of Ionic Interaction at the Muscarinic Receptor by A
Brit. J. Pharmacol. (1965), 25, 4-17. THE ROLE OF IONIC INTERACTION AT THE MUSCARINIC RECEPTOR BY A. S. V. BURGEN From the University Department ofPharmacology, Cambridge (Received August 27, 1964) It is commonly assumed by pharmacologists that the positively charged cationic group in such agonists as acetylcholine, noradrenaline and histamine is essential for their agonist action. The evidence in favour of this view is far from conclusive. Barlow & Hamilton (1962) examined the effect ofpH on the activity of nicotine in producing synaptic block of the rat phrenic nerve-diaphragm preparation. When compared with the quaternary nicotine N-methiodide the activity ofnicotine was related to thepH in the way to be expected if the nicotinium ion were more active than the non-protonated form, but neither the range ofpH values studied nor the precision of the results were sufficient to establish the activity (if any) of the non-protonated nicotine relative to the nicotinium ion. Hamilton (1963) has shown a similar effect of pH in the action of nicotine on the frog rectus abdominus muscle, but in this case also the activity of the non-protonated form was not established. Ariens, Simonis & van Rossum (1964) were unable to discover any clear relationship between pH and the action of arecoline on the frog rectus, but did not extend their study over a wide enough pH range to obtain decisive results. Rocha e Silva (1961) examined the effect ofpH on the action of histamine. There was a clear pH-dependence below pH 7 but the action was independent of pH above 7. -

A Pharmacological Analysis of Agonist and Tissue Dependence
Br. J. Pharmacol. (1992), 105, 643-652 .-) Macmillan Press Ltd, 1992 Inhibition of endothelium-dependent vasorelaxation by arginine analogues: a pharmacological analysis of agonist and tissue dependence 1G.R. Martin, M.L. Bolofo & H. Giles Analytical Pharmacology Group, Biochemical Sciences, Wellcome Research Laboratories, Beckenham, Kent BR3 3BS 1 Isolated rings of rabbit external jugular vein (RbJV) and rat thoracic aorta (RA) were used to study the effect of the NO synthase inhibitor L-NG-nitroarginine methyl ester (L-NAME) on muscarinic and 5- hydroxytryptamine (5-HT) receptor-stimulated, endothelium-dependent vascular relaxations. 2 In RbJV relaxations produced by the endothelial 5-HT receptor agonist a-methyl-5-HT were potently and non-surmountably inhibited by L-NAME (10pM), whereas acetylcholine relaxations in this tissue were unaffected by this concentration of inhibitor. By contrast, acetylcholine relaxations in RA were virtually abolished by 1O0 M L-NAME. In each case an equivalent concentration of D-NAME was without effect on agonist-induced relaxations. 3 The different effect of L-NAME on acetylcholine relaxations in RbJV and RA was not due to muscarinic receptor differences. Affinity estimates for acetylcholine (pKA = 6.12 + 0.09; 6.09 + 0.08 respectively) and for 4-diphenyl-acetoxy-N-methylpiperidine methobromide (4-DAMP, pKB = 9.01 + 0.12; 9.24 + 0.16 respectively) indicated that the receptors in both tissues belong to the same M3 class. Tissue differences resulting from the release of a cyclo-oxygenase product or a. glibenclamide-sensitive K+-channel-linked hyperpolarizing factor were also ruled out by selective inhibition of these pathways. 4 When phenoxybenzamine was used to reduce the efficacy of acetylcholine in RbJV so that it behaved as a partial agonist in this tissue, L-NAME (10pM) now produced non-surmountable inhibition of relax- ation responses. -

February 2019 Director's Report
TABLE OF CONTENTS RESEARCH HIGHLIGHTS…………….………………………………………………………. 1 GRANTEE HONORS AND AWARDS…………………………………………………………. 36 STAFF HONORS AND AWARDS…………………………………………………………….... 37 STAFF CHANGES…………………………………………………………………………….…. 43 IN MEMORIAM………………………………………………………………………………….. 48 RESEARCH FINDINGS BASIC AND BEHAVIORAL RESEARCH Adolescent Exposure To Δ9-Tetrahydrocannabinol Alters the Transcriptional Trajectory and Dendritic Architecture of Prefrontal Pyramidal Neurons Michael L. Miller, Benjamin Chadwick, Dara L. Dickstein, Immanuel Purushothaman, Gabor Egervari, Tanni Rahman, Chloe Tessereau, Patrick R. Hof, Panos Roussos, Li Shen, Mark G. Baxter, Yasmin L. Hurd; Mol. Psychiatry 2018. Neuronal circuits within the prefrontal cortex (PFC) mediate higher cognitive functions and emotional regulation that are disrupted in psychiatric disorders. The PFC undergoes significant maturation during adolescence, a period when cannabis use in humans has been linked to subsequent vulnerability to psychiatric disorders such as addiction and schizophrenia. Here, the authors investigated in a rat model the effects of adolescent exposure to Δ9-tetrahydrocannabinol (THC), a psychoactive component of cannabis, on the morphological architecture and transcriptional profile of layer III pyramidal neurons-using cell type- and layer-specific high- resolution microscopy, laser capture microdissection and next-generation RNA-sequencing. The results confirmed known normal expansions in basal dendritic arborization and dendritic spine pruning during the transition from late -

Biocompatible Choline Iodide Catalysts for Green Living Radical Polymerization of Functional Polymers
This document is downloaded from DR‑NTU (https://dr.ntu.edu.sg) Nanyang Technological University, Singapore. Biocompatible choline iodide catalysts for green living radical polymerization of functional polymers Wang, Chen‑Gang; Hanindita, Fiona; Goto, Atsushi 2018 Wang, C.‑G., Hanindita, F., & Goto, A. (2018). Biocompatible choline iodide catalysts for green living radical polymerization of functional polymers. ACS Macro Letters, 7(2), 263‑268. doi:10.1021/acsmacrolett.8b00026 https://hdl.handle.net/10356/105875 https://doi.org/10.1021/acsmacrolett.8b00026 © 2018 American Chemical Society. This document is the Accepted Manuscript version of a Published Work that appeared in final form in ACS Macro Letters, copyright © American Chemical Society after peer review and technical editing by the publisher. To access the final edited and published work see https://doi.org/10.1021/acsmacrolett.8b00026 Downloaded on 30 Sep 2021 00:34:09 SGT Biocompatible Choline Iodide Catalysts for Green Living Radical Polymerization of Functional Polymers Chen‐Gang Wang, Fiona Hanindita, and Atsushi Goto* Division of Chemistry and Biological Chemistry, School of Physical and Mathematical Sciences, Nanyang Techno‐ logical University, 21 Nanyang Link, 637371 Singapore ABSTRACT: Herein, non‐toxic and metabolizable choline iodide analogues, including choline iodide, acetylcholine iodide, and butyrylcholine iodide, were successfully utilized as novel catalysts for “green” living radical polymerization (LRP). Through the combination of several green solvents (ethyl lactate, ethanol, and water), this green LRP process yielded low‐ polydispersity hydrophobic, hydrophilic, zwitterionic, and water‐soluble biocompatible polymethacrylates and polyacry‐ lates with high monomer conversions. Well‐defined hydrophobic‐hydrophilic and hydrophilic‐hydrophilic block copoly‐ mers were also synthesized. -
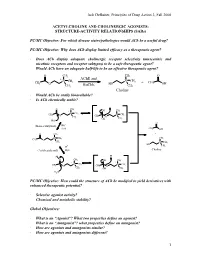
Jack Deruiter, Principles of Drug Action 2, Fall 2000 1 ACETYLCHOLINE and CHOLINERGIC AGONISTS: STRUCTURE-ACTIVITY RELATIONSHIPS
Jack DeRuiter, Principles of Drug Action 2, Fall 2000 ACETYLCHOLINE AND CHOLINERGIC AGONISTS: STRUCTURE-ACTIVITY RELATIONSHIPS (SARs) PC/MC Objective: For which disease states/pathologies would ACh be a useful drug? PC/MC Objective: Why does ACh display limited efficacy as a therapeutic agent? · Does ACh display adequate cholinergic receptor selectivity (muscarinic and nicotinic receptors and receptor subtypes) to be a safe therapeutic agent? · Would ACh have an adequate half-life to be an effective therapeutic agent? O CH3 CH3 O + AChE and + CH CH N CH3 N 3 + CH 3 O BuChE HO 3 OH CH3 CH3 Choline · Would ACh be orally bioavailable? · Is ACh chemically stable? O CH3 + OH CH3 + N CH3 CH3 N CH3 O CH3 CH3 O OH CH3 HO- O - (Base-catalyzed) HO H2O CH3 OH O CH3 + + N CH CH CH 3 3 3 O + CH3 N CH3 HO H+ CH3 (Acid-catalyzed) H2O Choline + H+ H O CH3 OH CH3 + + N CH3 N CH3 CH3 O CH3 CH O 3 OH CH3 H2O PC/MC Objective: How could the structure of ACh be modified to yield derivatives with enhanced therapeutic potential? · Selective agonist activity? · Chemical and metabolic stability? Global Objectives: · What is an “Agonist”? What two properties define an agonist? · What is an “Antagonist”? what properties define an antagonist? · How are agonists and antagonists similar? · How are agonists and antagonists different? 1 Jack DeRuiter, Principles of Drug Action 2, Fall 2000 MC Objective: Which ACh structural features appear to be required or at least contribute to cholinergic receptor binding. O CH3 + N CH3 CH3 O CH3 Ester Quaternary Ammonium MC Objective: -

Mitigation Potential of Selenium Nanoparticles and Riboflavin Against
www.nature.com/scientificreports OPEN Mitigation potential of selenium nanoparticles and ribofavin against arsenic and elevated temperature stress in Pangasianodon hypophthalmus Neeraj Kumar1*, Sanjay Kumar Gupta2, Nitish Kumar Chandan3, Shashi Bhushan4, Dilip Kumar Singh4, Paritosh Kumar1, Prem Kumar5, Goraksha C. Wakchaure1 & Narendra Pratap Singh1 Climate change impact has disturbed the rainfall pattern worsening the problems of water availability in the aquatic ecosystem of India and other parts of the world. Arsenic pollution, mainly through excessive use of groundwater and other anthropogenic activities, is aggravating in many parts of the world, particularly in South Asia. We evaluated the efcacy of selenium nanoparticles (Se-NPs) and ribofavin (RF) to ameliorate the adverse impacts of elevated temperature and arsenic pollution on growth, anti-oxidative status and immuno-modulation in Pangasianodon hypophthalmus. Se-NPs were synthesized using fsh gill employing green synthesis method. Four diets i.e., Se-NPs (0 mg kg−1) + RF (0 mg kg−1); Se-NPs (0.5 mg kg−1) + RF (5 mg kg−1); Se-NPs (0.5 mg kg−1) + RF (10 mg kg−1); and Se-NPs (0.5 mg kg−1) + RF (15 mg kg−1) were given in triplicate in a completely −1 randomized block design. The fsh were treated in arsenic (1/10th of LC50, 2.68 mg L ) and high temperature (34 °C). Supplementation of the Se-NPs and RF in the diets signifcantly (p < 0.01) enhanced growth performance (weight gain, feed efciency ratio, protein efciency ratio, and specifc growth rate), anti-oxidative status and immunity of the fsh. Nitroblue tetrazolium (NBT), total immunoglobulin, myeloperoxidase and globulin enhanced (p < 0.01) with supplementation (Se-NPs + RF) whereas, albumin and albumin globulin (A:G) ratio (p < 0.01) reduced.