The NBS-LRR Architectures of Plant R-Proteins and Metazoan Nlrs Evolved in Independent Events
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

A Novel LRRK2 Variant P.G2294R in the WD40 Domain Identified in Familial Parkinson's Disease Affects LRRK2 Protein Levels
International Journal of Molecular Sciences Article A Novel LRRK2 Variant p.G2294R in the WD40 Domain Identified in Familial Parkinson’s Disease Affects LRRK2 Protein Levels Jun Ogata 1, Kentaro Hirao 2, Kenya Nishioka 3 , Arisa Hayashida 3, Yuanzhe Li 3, Hiroyo Yoshino 4, Soichiro Shimizu 2, Nobutaka Hattori 1,3,4 and Yuzuru Imai 1,* 1 Department of Research for Parkinson’s Disease, Juntendo University Graduate School of Medicine, Tokyo 113-8421, Japan; [email protected] (J.O.); [email protected] (N.H.) 2 Department of Geriatric Medicine, Tokyo Medical University, 6-7-1 Nishishinjuku, Shinjuku-ku, Tokyo 160-0023, Japan; [email protected] (K.H.); [email protected] (S.S.) 3 Department of Neurology, Juntendo University School of Medicine, 2-1-1 Hongo, Bunkyo-ku, Tokyo 113-8421, Japan; [email protected] (K.N.); [email protected] (A.H.); [email protected] (Y.L.) 4 Research Institute for Diseases of Old Age, Graduate School of Medicine, Juntendo University, 2-1-1 Hongo, Bunkyo-ku, Tokyo 113-8421, Japan; [email protected] * Correspondence: [email protected]; Tel.: +81-3-6801-8332 Abstract: Leucine-rich repeat kinase 2 (LRRK2) is a major causative gene of late-onset familial Parkin- son’s disease (PD). The suppression of kinase activity is believed to confer neuroprotection, as most pathogenic variants of LRRK2 associated with PD exhibit increased kinase activity. We herein report a novel LRRK2 variant—p.G2294R—located in the WD40 domain, detected through targeted gene- Citation: Ogata, J.; Hirao, K.; panel screening in a patient with familial PD. -
Supplementary Table 2 Supplementary Table 1
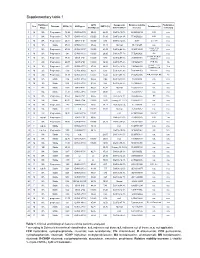
Supplementary table 1 Rai/ Binet IGHV Cytogenetic Relative viability Fludarabine- Sex Outcome CD38 (%) IGHV gene ZAP70 (%) Treatment (s) Stage identity (%) abnormalities* increase refractory 1 M 0/A Progressive 14,90 IGHV3-64*05 99,65 28,20 Del17p 18.0% 62,58322819 FCR n.a. 2 F 0/A Progressive 78,77 IGHV3-48*03 100,00 51,90 Del17p 24.8% 77,88052021 FCR n.a. 3 M 0/A Progressive 29,81 IGHV4-b*01 100,00 9,10 Del17p 12.0% 36,48 Len, Chl n.a. 4 M 1/A Stable 97,04 IGHV3-21*01 97,22 18,11 Normal 85,4191657 n.a. n.a. Chl+O, PCR, 5 F 0/A Progressive 87,00 IGHV4-39*07 100,00 43,20 Del13q 68.3% 35,23314039 n.a. HDMP+R 6 M 0/A Progressive 1,81 IGHV3-43*01 100,00 20,90 Del13q 77.7% 57,52490626 Chl n.a. Chl, FR, R-CHOP, 7 M 0/A Progressive 97,80 IGHV1-3*01 100,00 9,80 Del17p 88.5% 48,57389901 n.a. HDMP+R 8 F 2/B Progressive 69,07 IGHV5-a*03 100,00 16,50 Del17p 77.2% 107,9656878 FCR, BA No R-CHOP, FCR, 9 M 1/A Progressive 2,13 IGHV3-23*01 97,22 29,80 Del11q 16.3% 134,5866919 Yes Flavopiridol, BA 10 M 2/A Progressive 0,36 IGHV3-30*02 92,01 0,38 Del13q 81.9% 78,91844953 Unknown n.a. 11 M 2/B Progressive 15,17 IGHV3-20*01 100,00 13,20 Del11q 95.3% 75,52880995 FCR, R-CHOP, BR No 12 M 0/A Stable 0,14 IGHV3-30*02 90,62 7,40 Del13q 13.0% 13,0939004 n.a. -

ATP-Binding and Hydrolysis in Inflammasome Activation
molecules Review ATP-Binding and Hydrolysis in Inflammasome Activation Christina F. Sandall, Bjoern K. Ziehr and Justin A. MacDonald * Department of Biochemistry & Molecular Biology, Cumming School of Medicine, University of Calgary, 3280 Hospital Drive NW, Calgary, AB T2N 4Z6, Canada; [email protected] (C.F.S.); [email protected] (B.K.Z.) * Correspondence: [email protected]; Tel.: +1-403-210-8433 Academic Editor: Massimo Bertinaria Received: 15 September 2020; Accepted: 3 October 2020; Published: 7 October 2020 Abstract: The prototypical model for NOD-like receptor (NLR) inflammasome assembly includes nucleotide-dependent activation of the NLR downstream of pathogen- or danger-associated molecular pattern (PAMP or DAMP) recognition, followed by nucleation of hetero-oligomeric platforms that lie upstream of inflammatory responses associated with innate immunity. As members of the STAND ATPases, the NLRs are generally thought to share a similar model of ATP-dependent activation and effect. However, recent observations have challenged this paradigm to reveal novel and complex biochemical processes to discern NLRs from other STAND proteins. In this review, we highlight past findings that identify the regulatory importance of conserved ATP-binding and hydrolysis motifs within the nucleotide-binding NACHT domain of NLRs and explore recent breakthroughs that generate connections between NLR protein structure and function. Indeed, newly deposited NLR structures for NLRC4 and NLRP3 have provided unique perspectives on the ATP-dependency of inflammasome activation. Novel molecular dynamic simulations of NLRP3 examined the active site of ADP- and ATP-bound models. The findings support distinctions in nucleotide-binding domain topology with occupancy of ATP or ADP that are in turn disseminated on to the global protein structure. -

NLR Members in Inflammation-Associated
Cellular & Molecular Immunology (2017) 14, 403–405 & 2017 CSI and USTC All rights reserved 2042-0226/17 $32.00 www.nature.com/cmi RESEARCH HIGHTLIGHT NLR members in inflammation-associated carcinogenesis Ha Zhu1,2 and Xuetao Cao1,2,3 Cellular & Molecular Immunology (2017) 14, 403–405; doi:10.1038/cmi.2017.14; published online 3 April 2017 hronic inflammation is regarded as an impor- nucleotide-binding and oligomerization domain IL-2,8 and NAIP was found to regulate the STAT3 Ctant factor in cancer progression. In addition (NOD)-like receptors (NLRs). TLRs and CLRs are pathway independent of inflammasome formation.9 to the immune surveillance function in the early located in the plasma membranes, whereas RLRs, The AOM/DSS model is the most popular model stage of tumorigenesis, inflammation is also known ALRs and NLRs are intracellular PRRs.3 Unlike used to study the function of NLRs in fl fl as one of the hallmarks of cancer and can supply other families that have been shown to bind their in ammation-associated carcinogenesis. In amma- the tumor microenvironment with bioactive mole- specific cognate ligands, the distinct ligands for somes initiated by NLRs or AIM2 have been widely cules and favor the development of other hallmarks NLRs are still unknown. In fact, mounting evidence reported to participate in the maintenance of 10,11 Nlrp3 Nlrp6 of cancer, such as genetic instability and angiogen- suggests that NLRs function as cytoplasmic sensors intestinal homeostasis. -/-, -/-, Nlrc4 Nlrp1 Nlrx1 Nlrp12 esis. Moreover, inflammation contributes to the and participate in modulating TLR, RLR and CLR -/-, -/-, -/- and -/- mice are 4 more susceptible to AOM/DSS-induced colorectal changing tumor microenvironment by altering signaling pathways. -

Apoptosis Ligand-Induced Enhanced Resistance to Fas/Fas
Theileria parva-Transformed T Cells Show Enhanced Resistance to Fas/Fas Ligand-Induced Apoptosis This information is current as Peter Küenzi, Pascal Schneider and Dirk A. E. Dobbelaere of October 2, 2021. J Immunol 2003; 171:1224-1231; ; doi: 10.4049/jimmunol.171.3.1224 http://www.jimmunol.org/content/171/3/1224 Downloaded from References This article cites 69 articles, 29 of which you can access for free at: http://www.jimmunol.org/content/171/3/1224.full#ref-list-1 Why The JI? Submit online. http://www.jimmunol.org/ • Rapid Reviews! 30 days* from submission to initial decision • No Triage! Every submission reviewed by practicing scientists • Fast Publication! 4 weeks from acceptance to publication *average by guest on October 2, 2021 Subscription Information about subscribing to The Journal of Immunology is online at: http://jimmunol.org/subscription Permissions Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Email Alerts Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts The Journal of Immunology is published twice each month by The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2003 by The American Association of Immunologists All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. The Journal of Immunology Theileria parva-Transformed T Cells Show Enhanced Resistance to Fas/Fas Ligand-Induced Apoptosis1 Peter Ku¨enzi,2* Pascal Schneider,† and Dirk A. E. Dobbelaere3* Lymphocyte homeostasis is regulated by mechanisms that control lymphocyte proliferation and apoptosis. Activation-induced cell death is mediated by the expression of death ligands and receptors, which, when triggered, activate an apoptotic cascade. -

De Novo PHIP Predicted Deleterious Variants Are Associated with Developmental Delay, Intellectual Disability, Obesity, and Dysmorphic Features
Downloaded from molecularcasestudies.cshlp.org on October 4, 2021 - Published by Cold Spring Harbor Laboratory Press De novo PHIP predicted deleterious variants are associated with developmental delay, intellectual disability, obesity, and dysmorphic features Running title: Case study of two patients with PHIP variants Emily Webster1, Megan T. Cho2, Nora Alexander2, Sonal Desai3, Sakkubai Naidu3, Mir Reza Bekheirnia4, Andrea Lewis4, Kyle Retterer2, Jane Juusola2, Wendy K. Chung1,5 1Department of Pediatrics, Columbia University Medical Center, New York, NY, USA 2GeneDx, Gaithersburg, MD, USA 3Kennedy Krieger Institute, Baltimore, MD, USA 4Department of Molecular and Human Genetics, Baylor College of Medicine, Houston, TX, USA 5Department of Medicine, Columbia University Medical Center, New York, NY, USA Correspondence: Wendy K. Chung, Columbia University Medical Center, 1150 St. Nicholas Avenue, New York, NY 10032, USA, Tel: +1 212 851 5313, Fax: +1 212 851 5306, [email protected] Running title: PHIP variants cause developmental delay and obesity 1 Downloaded from molecularcasestudies.cshlp.org on October 4, 2021 - Published by Cold Spring Harbor Laboratory Press Abstract Using whole exome sequencing, we have identified novel de novo heterozygous Pleckstrin homology domain-interacting protein (PHIP) variants that are predicted to be deleterious, including a frameshift deletion, in two unrelated patients with common clinical features of developmental delay, intellectual disability, anxiety, hypotonia, poor balance, obesity, and dysmorphic features. A nonsense mutation in PHIP has previously been associated with similar clinical features. Patients with microdeletions of 6q14.1 including PHIP have a similar phenotype of developmental delay, intellectual disability, hypotonia, and obesity, suggesting that the phenotype of our patients is a result of loss-of-function mutations. -

Techniques for Immune Function Analysis Application Handbook 1St Edition
Techniques for Immune Function Analysis Application Handbook 1st Edition BD Biosciences For additional information please access the Immune Function Homepage at www.bdbiosciences.com/immune_function For Research Use Only. Not for use in diagnostic or therapeutic procedures. Purchase does not include or carry any right to resell or transfer this product either as a stand-alone product or as a component of another product. Any use of this product other than the permitted use without the express written authorization of Becton Dickinson and Company is strictly prohibited. All applications are either tested in-house or reported in the literature. See Technical Data Sheets for details. BD, BD Logo and all other trademarks are the property of Becton, Dickinson and Company. ©2003 BD Table of Contents Preface . 4 Chapter 1: Immunofluorescent Staining of Cell Surface Molecules for Flow Cytometric Analysis . 9 Chapter 2: BD™ Cytometric Bead Array (CBA) Multiplexing Assays . 35 Chapter 3: BD™ DimerX MHC:Ig Proteins for the Analysis of Antigen-specific T Cells. 51 Chapter 4: Immunofluorescent Staining of Intracellular Molecules for Flow Cytometric Analysis . 61 Chapter 5: BD FastImmune™ Cytokine Flow Cytometry. 85 Chapter 6: BD™ ELISPOT Assays for Cells That Secrete Biological Response Modifiers . 109 Chapter 7: ELISA for Specifically Measuring the Levels of Cytokines, Chemokines, Inflammatory Mediators and their Receptors . 125 Chapter 8: BD OptEIA™ ELISA Sets and Kits for Quantitation of Analytes in Serum, Plasma, and Cell Culture Supernatants. 143 Chapter 9: BrdU Staining and Multiparameter Flow Cytometric Analysis of the Cell Cycle . 155 Chapter 10: Cell-based Assays for Biological Response Modifiers . 177 Chapter 11: BD RiboQuant™ Multi-Probe RNase Protection Assay System . -

Molecular Genetics of Spinal Muscular Atrophy: Contribution of the NAIP Gene to Clinical Severity
Kobe J. Med. Sci. 48, 25/31 February 2002 Molecular Genetics of Spinal Muscular Atrophy: Contribution of the NAIP Gene to Clinical Severity TOMOKO AKUTSU1*, HISAHIDE NISHIO2, KIMIAKI SUMINO2, YASUHIRO TAKESHIMA1, SYUICHI TSUNEISHI1, HIROKO WADA1, SATOSHI TAKADA3, MASAFUMI MATSUO1, 4, and HAJIME NAKAMURA1 Division of Pediatrics, Department of Development and Aging1, Division of Public Health, Department of Environmental Health and Safety2, Kobe University Graduate School of Medicine Faculty of Health Science, Kobe University School of Medicine, Kobe 654-0142, Japan3 Division of Molecular Medicine, Department of International and Environmental Medical Sciences, Kobe University Graduate School of Medicine4 Received 5 February 2002/ Accepted 19 February 2002 Key words: spinal muscular atrophy; the SMN1 gene; the NAIP gene; the p44t gene Spinal muscular atrophy (SMA) is one of the most common autosomal recessive disorders characterized by degeneration of anterior horn cells in the spinal cord, and leads to progressive muscular weakness and atrophy. At least three SMA-related genes have been identified: SMN1, NAIP and p44t. We analyzed these genes in 32 SMA patients and found that the SMN1 gene was deleted in 30 of 32 patients (94 %), irrespective of clinical type. The NAIP gene was deleted in 6 patients and its deletion rate was higher in type I patients than that in typeⅡ or Ⅲ. Further, in type I patients lacking the NAIP gene, deterioration in their respiratory function is more rapid than in those type I patients retaining the NAIP gene. Since complete p44t deletion was observed in only 3 patients, the correlation between the p44t deletion and severity of SMA remained ambiguous. -

The Role of Tlrs, Nlrs, and Rlrs in Mucosal Innate Immunity and Homeostasis
nature publishing group REVIEW See ARTICLE page 29 The role of TLRs, NLRs, and RLRs in mucosal innate immunity and homeostasis E C L a v e l l e 1 , C M u r p h y 2 , L A J O ’ N e i l l 2 a n d E M C r e a g h 2 The mucosal surfaces of the gastrointestinal tract are continually exposed to an enormous antigenic load of microbial and dietary origin, yet homeostasis is maintained. Pattern recognition molecules (PRMs) have a key role in maintaining the integrity of the epithelial barrier and in promoting maturation of the mucosal immune system. Commensal bacteria modulate the expression of a broad range of genes involved in maintaining epithelial integrity, inflammatory responses, and production of antimicrobial peptides. Mice deficient in PRMs can develop intestinal inflammation, which is dependent on the microbiota, and in humans, PRM polymorphisms are associated with exacerbated inflammatory bowel disease. Innate immune responses and epithelial barrier function are regulated by PRM-induced signaling at multiple levels, from the selective expression of receptors on mucosal cells or compartments to the expression of negative regulators. Here, we describe recent advances in our understanding of innate signaling pathways, particularly by Toll-like receptors and nucleotide-binding domain and leucine-rich repeat containing receptors at mucosal surfaces. INTRODUCTION mucosal immune compartment, which comprises the lamina Activation of the innate immune system is triggered when patho- propria, Peyer ’ s patches, and mesenteric lymph nodes.4 The gen-associated molecular patterns or endogenous damage-asso- ability to contain the microbiota in this region is dependent on ciated molecular patterns engage pattern recognition molecules PRMs, as a deficiency in TLR signaling results in the increased (PRMs) on cells including epithelial cells, macrophages, and den- uptake of bacteria into the spleens of mice. -

Coronin 3 Involvement in F-Actin-Dependent Processes at the Cell Cortex
Coronin 3 involvement in F-actin-dependent processes at the cell cortex Author Rosentreter, Andre, Hofmann, Andreas, Xavier, Charles-Peter, Stumpf, Maria, Noegel, Angelika A, Clemen, Christoph S Published 2007 Journal Title Experimental Cell Research DOI https://doi.org/10.1016/j.yexcr.2006.12.015 Copyright Statement © 2007 Elsevier. This is the author-manuscript version of this paper. Reproduced in accordance with the copyright policy of the publisher. Please refer to the journal's website for access to the definitive, published version. Downloaded from http://hdl.handle.net/10072/14982 Griffith Research Online https://research-repository.griffith.edu.au Elsevier Editorial System(tm) for Experimental Cell Research Manuscript Draft Manuscript Number: Title: Coronin 3 involvement in F-actin dependent processes at the cell cortex Article Type: Research Article Section/Category: Keywords: Coro1C; CRNN4; WD40-repeat; F-actin; cell motility; cytoskeleton; siRNA Corresponding Author: Dr. Christoph Stephan Clemen, Corresponding Author's Institution: Institute of Biochemistry I First Author: André Rosentreter Order of Authors: André Rosentreter; Andreas Hofmann; Charles-Peter Xavier; Maria Stumpf; Angelika Anna Noegel; Christoph Stephan Clemen Manuscript Region of Origin: Abstract: The actin interaction of coronin 3 has been mainly documented by in vitro experiments. Here, we discuss coronin 3 properties in the light of new structural information and focus on assays that reflect in vivo roles of coronin 3 and its impact on F-actin associated functions. Using GFP-tagged coronin 3 fusion proteins and RNAi silencing we show that coronin 3 has roles in wound healing, protrusion formation, cell proliferation, cytokinesis, endocytosis, axonal growth, and secretion. -

NOD1 Activators Link Innate Immunity to Insulin Resistance
ORIGINAL ARTICLE NOD1 Activators Link Innate Immunity to Insulin Resistance Jonathan D. Schertzer,1,2 Akhilesh K. Tamrakar,1 Joao G. Magalhães,3 Sandra Pereira,4 Philip J. Bilan,1 Morgan D. Fullerton,2 Zhi Liu,1 Gregory R. Steinberg,2 Adria Giacca,4 Dana J. Philpott,3 and Amira Klip1 OBJECTIVE—Insulin resistance associates with chronic inflam- mation, and participatory elements of the immune system are emerging. We hypothesized that bacterial elements acting on nsulin resistance is a major predictor and leading distinct intracellular pattern recognition receptors of the innate cause of type 2 diabetes (1). Intricate links between immune system, such as bacterial peptidoglycan (PGN) acting on metabolic and immune responses underlie the dis- nucleotide oligomerization domain (NOD) proteins, contribute to ease development (2) and a chronic, low-level in- insulin resistance. I flammation has been associated with insulin resistance RESEARCH DESIGN AND METHODS—Metabolic and in- (3,4). Innate and adaptive immune systems have been flammatory properties were assessed in wild-type (WT) and implicated in the proinflammatory responses and have 2/2 NOD1/2 double knockout mice fed a high-fat diet (HFD) for emerged as critical factors in the manifestation of insulin 16 weeks. Insulin resistance was measured by hyperinsulinemic resistance (5–7). It is paramount to define the specificim- euglycemic clamps in mice injected with mimetics of meso- fl diaminopimelic acid–containing PGN or the minimal bioactive mune elements that mediate in ammation-induced meta- PGN motif, which activate NOD1 and NOD2, respectively. Systemic bolic alterations. This may yield therapeutic approaches and tissue-specificinflammation was assessed using enzyme-linked to combat insulin resistance by divorcing metabolic and immunosorbent assays in NOD ligand–injected mice. -

Coronin7 Regulates WASP and SCAR Through CRIB Mediated Interaction
www.nature.com/scientificreports OPEN Coronin7 regulates WASP and SCAR through CRIB mediated interaction with Rac proteins Received: 12 May 2015 1,† 1 1 1 Accepted: 28 August 2015 Karthic Swaminathan , Maria Stumpf , Rolf Müller , Anna-Carolin Horn , 1 1 2 3 Published: 28 September 2015 Julia Schmidbauer , Ludwig Eichinger , Annette Müller-Taubenberger , Jan Faix & Angelika A. Noegel1 Coronin7 (CRN7) stabilizes F-actin and is a regulator of processes associated with the actin cytoskeleton. Its loss leads to defects in phagocytosis, motility and development. It harbors a CRIB (Cdc42- and Rac-interactive binding) domain in each of its WD repeat domains which bind to Rac GTPases preferably in their GDP-loaded forms. Expression of wild type CRN7 in CRN7 deficient cells rescued these defects, whereas proteins with mutations in the CRIB motifs which were associated with altered Rac binding were effective to varying degrees. The presence of one functional CRIB was sufficient to reestablish phagocytosis, cell motility and development. Furthermore, by molecular modeling and mutational analysis we identified the contact regions between CRN7 and the GTPases. We also identified WASP, SCAR and PAKa as downstream effectors in phagocytosis, development and cell surface adhesion, respectively, since ectopic expression rescued these functions. Coronins are a large family of evolutionary conserved proteins. They consist of a WD (Tryptophane- Aspartate)-repeat domain containing seven repeats that form a seven-bladed β -propeller structur- ally resembling the Gβ subunit of the heterotrimeric G-proteins. This region is followed by a unique region and a C-terminal coiled coil region which mediates oligomerization. Coronins play roles in actin cytoskeleton-associated processes, in signal transduction, in endosomal trafficking, survival of pathogenic bacteria in macrophages and homeostatic T cell signalling1–3.