Congenital Corneal Opacities KK Nischal 1327
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

National Study of Microphthalmia, Anophthalmia, and Coloboma (MAC
16 ORIGINAL ARTICLE J Med Genet: first published as 10.1136/jmg.39.1.16 on 1 January 2002. Downloaded from National study of microphthalmia, anophthalmia, and coloboma (MAC) in Scotland: investigation of genetic aetiology D Morrison, D FitzPatrick, I Hanson, K Williamson, V van Heyningen, B Fleck, I Jones, J Chalmers, H Campbell ............................................................................................................................. J Med Genet 2002;39:16–22 We report an epidemiological and genetic study attempting complete ascertainment of subjects with microphthalmia, anophthalmia, and coloboma (MAC) born in Scotland during a 16 year period beginning on 1 January 1981. A total of 198 cases were confirmed giving a minimum live birth preva- lence of 19 per 100 000. One hundred and twenty-two MAC cases (61.6%) from 115 different fami- See end of article for lies were clinically examined and detailed pregnancy, medical, and family histories obtained. A authors’ affiliations simple, rational, and apparently robust classification of the eye phenotype was developed based on ....................... the presence or absence of a defect in closure of the optic (choroidal) fissure. A total of 85/122 Correspondence to: (69.7%) of cases had optic fissure closure defects (OFCD), 12/122 (9.8%) had non-OFCD, and Dr D FitzPatrick, MRC 25/122 (20.5%) had defects that were unclassifiable owing to the severity of the corneal or anterior Human Genetics Unit, chamber abnormality. Segregation analysis assuming single and multiple incomplete ascertainment, Western General Hospital, respectively, returned a sib recurrence risk of 6% and 10% in the whole group and 8.1% and 13.3% Edinburgh EH4 2XU, UK; in the OFCD subgroup. -

Corneal Ectasia
Corneal Ectasia Secretary for Quality of Care Anne L. Coleman, MD, PhD Academy Staff Nicholas P. Emptage, MAE Nancy Collins, RN, MPH Doris Mizuiri Jessica Ravetto Flora C. Lum, MD Medical Editor: Susan Garratt Design: Socorro Soberano Approved by: Board of Trustees September 21, 2013 Copyright © 2013 American Academy of Ophthalmology® All rights reserved AMERICAN ACADEMY OF OPHTHALMOLOGY and PREFERRED PRACTICE PATTERN are registered trademarks of the American Academy of Ophthalmology. All other trademarks are the property of their respective owners. This document should be cited as follows: American Academy of Ophthalmology Cornea/External Disease Panel. Preferred Practice Pattern® Guidelines. Corneal Ectasia. San Francisco, CA: American Academy of Ophthalmology; 2013. Available at: www.aao.org/ppp. Preferred Practice Pattern® guidelines are developed by the Academy’s H. Dunbar Hoskins Jr., MD Center for Quality Eye Care without any external financial support. Authors and reviewers of the guidelines are volunteers and do not receive any financial compensation for their contributions to the documents. The guidelines are externally reviewed by experts and stakeholders before publication. Corneal Ectasia PPP CORNEA/EXTERNAL DISEASE PREFERRED PRACTICE PATTERN DEVELOPMENT PROCESS AND PARTICIPANTS The Cornea/External Disease Preferred Practice Pattern® Panel members wrote the Corneal Ectasia Preferred Practice Pattern® guidelines (“PPP”). The PPP Panel members discussed and reviewed successive drafts of the document, meeting in person twice and conducting other review by e-mail discussion, to develop a consensus over the final version of the document. Cornea/External Disease Preferred Practice Pattern Panel 2012–2013 Robert S. Feder, MD, Co-chair Stephen D. McLeod, MD, Co-chair Esen K. -

Infantile Aphakia and Successful Fitting of Pediatric Contact Lenses; a Case Presentation Authors: Virji N, Patel A, Libassi D
Infantile aphakia and successful fitting of pediatric contact lenses; a case presentation Authors: Virji N, Patel A, Libassi D An eleven month old male presents with bilateral aphakia secondary to congenital cataracts. The patient is currently successfully wearing B&L Silsoft Pediatric contact lenses, with good prognosis for vision in both eyes. I. Case History -Patient demographics: African American male, DOB 8/18/2009 -Chief complaint: patient presents with bilateral aphakia secondary to bilateral congenital cataract extraction -Ocular, medical history: S/P CE with anterior vitrectomy OD 09/22/2009, followed by OS 09/29/09. (+) squinting, rubs eyes, light sensitivity -Medications: none -Other salient information: patient has been seen by SUNY Contact Lens clinic since 2 months old, 10/14/2009 II. Pertinent findings -Clinical: Keratometry readings 41.00/41.25 @ 005 OD, 38.50/41.00 @ 046 Axial length, immeasurable Horizontal corneal diameter 8mm OD/OS Fundus exam WNL OU -Others: surgical dates: successful CE OU, September 2009 III. Differential diagnosis -Primary/leading: Idiopathic -Others: Posterior lenticonus, persistent hyperplastic primary vitreous, anterior segment dysgenesis, and posterior pole tumors, trauma, intrauterine infection (rubella), maternal hypoglycemia, trisomy (eg, Down, Edward, and Patau syndromes), myotonic dystrophy, infectious diseases (eg, toxoplasmosis, rubella, cytomegalovirus, and herpes simplex [TORCH]), and prematurity. (5) IV. Diagnosis and discussion -Elaborate on the condition: Bilateral infantile cataracts are one of the major treatable causes of visual impairment in children. (2) Hubel and Weisel’s research on the critical period of visual development determined that if infantile cataracts are removed within the critical period and appropriate correction is worn, vision is greatly improved. -

Whole Exome Sequencing Gene Package Vision Disorders, Version 6.1, 31-1-2020
Whole Exome Sequencing Gene package Vision disorders, version 6.1, 31-1-2020 Technical information DNA was enriched using Agilent SureSelect DNA + SureSelect OneSeq 300kb CNV Backbone + Human All Exon V7 capture and paired-end sequenced on the Illumina platform (outsourced). The aim is to obtain 10 Giga base pairs per exome with a mapped fraction of 0.99. The average coverage of the exome is ~50x. Duplicate and non-unique reads are excluded. Data are demultiplexed with bcl2fastq Conversion Software from Illumina. Reads are mapped to the genome using the BWA-MEM algorithm (reference: http://bio-bwa.sourceforge.net/). Variant detection is performed by the Genome Analysis Toolkit HaplotypeCaller (reference: http://www.broadinstitute.org/gatk/). The detected variants are filtered and annotated with Cartagenia software and classified with Alamut Visual. It is not excluded that pathogenic mutations are being missed using this technology. At this moment, there is not enough information about the sensitivity of this technique with respect to the detection of deletions and duplications of more than 5 nucleotides and of somatic mosaic mutations (all types of sequence changes). HGNC approved Phenotype description including OMIM phenotype ID(s) OMIM median depth % covered % covered % covered gene symbol gene ID >10x >20x >30x ABCA4 Cone-rod dystrophy 3, 604116 601691 94 100 100 97 Fundus flavimaculatus, 248200 {Macular degeneration, age-related, 2}, 153800 Retinal dystrophy, early-onset severe, 248200 Retinitis pigmentosa 19, 601718 Stargardt disease -

Intraocular Lenses and Spectacle Correction
MEDICAL POLICY POLICY TITLE INTRAOCULAR LENSES, SPECTACLE CORRECTION AND IRIS PROSTHESIS POLICY NUMBER MP-6.058 Original Issue Date (Created): 6/2/2020 Most Recent Review Date (Revised): 6/9/2020 Effective Date: 2/1/2021 POLICY PRODUCT VARIATIONS DESCRIPTION/BACKGROUND RATIONALE DEFINITIONS BENEFIT VARIATIONS DISCLAIMER CODING INFORMATION REFERENCES POLICY HISTORY I. POLICY Intraocular Lens Implant (IOL) Initial IOL Implant A standard monofocal intraocular lens (IOL) implant is medically necessary when the eye’s natural lens is absent including the following: Following cataract extraction Trauma to the eye which has damaged the lens Congenital cataract Congenital aphakia Lens subluxation/displacement A standard monofocal intraocular lens (IOL) implant is medically necessary for anisometropia of 3 diopters or greater, and uncorrectable vision with the use of glasses or contact lenses. Premium intraocular lens implants including but not limited to the following are not medically necessary for any indication, including aphakia, because each is intended to reduce the need for reading glasses. Presbyopia correcting IOL (e.g., Array® Model SA40, ReZoom™, AcrySof® ReStor®, TECNIS® Multifocal IOL, Tecnis Symfony and Tecnis SymfonyToric, TRULIGN, Toric IO, Crystalens Aspheric Optic™) Astigmatism correcting IOL (e.g., AcrySof IQ Toric IOL (Alcon) and Tecnis Toric Aspheric IOL) Phakic IOL (e.g., ARTISAN®, STAAR Visian ICL™) Replacement IOLs MEDICAL POLICY POLICY TITLE INTRAOCULAR LENSES, SPECTACLE CORRECTION AND IRIS PROSTHESIS POLICY NUMBER -

Insertion of Aqueous Shunt in Pedicatric Glaucoma
1/29/2018 Challenges of Insertion of Aqueous shunt in paediatric glaucoma Ahmed Elkarmouty MD, FRCS Moorfields Eye Hospital London, UK Classification • Primary Childhood Glaucoma • A- Primary Congenital Glaucoma (PCG) 1: 10,000–18,000 • B- Juvenile Open Angle Glaucoma (JOAG) (5-35 ys,)1 : 50,000. • Secondary Childhood Glaucoma • A- Glaucoma associated with non-acquired ocular anomalies • B- Glaucoma associated with non- acquired systemic disease or syndrome • C- Glaucoma associated with acquired condition • D- Glaucoma following Cataract surgery 1 1/29/2018 Glaucoma associated with non- acquired ocular anomalies • Conditions with predominantly ocular anomalies present at birth which may or may not be associated with systemic signs • Axenfeld Reiger anomaly • Peters anomaly • Ectropion Uvae • Congenital iris hypolplasia • Aniridia • Oculodermal melanocytosis • Posterior polymorphous dystrophy • Microphthalmos • Microcornea • Ectopia Lentis ( et pupillae) • Persistent foetus vasculopathy Glaucoma associated with non- acquired systemic disease or syndrome predominantly associated with known syndrome, systemic anomalies present at birth which may be associated with ocular signs • Down Syndrome • Connective tissue disorder: Marfan syndrome, Weill- Marchesiani syndrome, Stickler syndrome • Metabolic disorder : Homocystenuria, lowe syndrome, Mucoploysacchroidoses • Phacomatoses: Neurofibromatoses, Sturge Weber, Klipple-Trenaunay- weber syndrome, Rubenstein Taybi • Congenital Rubella 2 1/29/2018 Glaucoma associated with acquired condition Conditions -

Expanding the Phenotypic Spectrum of PAX6 Mutations: from Congenital Cataracts to Nystagmus
G C A T T A C G G C A T genes Article Expanding the Phenotypic Spectrum of PAX6 Mutations: From Congenital Cataracts to Nystagmus Maria Nieves-Moreno 1,* , Susana Noval 1 , Jesus Peralta 1, María Palomares-Bralo 2 , Angela del Pozo 3 , Sixto Garcia-Miñaur 4, Fernando Santos-Simarro 4 and Elena Vallespin 5 1 Department of Ophthalmology, Hospital Universitario La Paz, 28046 Madrid, Spain; [email protected] (S.N.); [email protected] (J.P.) 2 Department of Molecular Developmental Disorders, Medical and Molecular Genetics Institue (INGEMM) IdiPaz, CIBERER, Hospital Universitario La Paz, 28046 Madrid, Spain; [email protected] 3 Department of Bioinformatics, Medical and Molecular Genetics Institue (INGEMM) IdiPaz, CIBERER, Hospital Universitario La Paz, 28046 Madrid, Spain; [email protected] 4 Department of Clinical Genetics, Medical and Molecular Genetics Institue (INGEMM) IdiPaz, CIBERER, Hospital Universitario La Paz, 28046 Madrid, Spain; [email protected] (S.G.-M.); [email protected] (F.S.-S.) 5 Department of Molecular Ophthalmology, Medical and Molecular Genetics Institue (INGEMM) IdiPaz, CIBERER, Hospital Universitario La Paz, 28046 Madrid, Spain; [email protected] * Correspondence: [email protected] Abstract: Background: Congenital aniridia is a complex ocular disorder, usually associated with severe visual impairment, generally caused by mutations on the PAX6 gene. The clinical phenotype of PAX6 mutations is highly variable, making the genotype–phenotype correlations difficult to establish. Methods: we describe the phenotype of eight patients from seven unrelated families Citation: Nieves-Moreno, M.; Noval, with confirmed mutations in PAX6, and very different clinical manifestations. -

Ophthalmology
Ophthalmology Information for health professionals MEDICAL GENETIC TESTING FOR OPHTHALMOLOGY Recent technologies, in particularly Next Generation Sequencing (NGS), allows fast, accurate and valuable diagnostic tests. For Ophthalmology, CGC Genetics has an extensive list of medical genetic tests with clinical integration of results by our Medical Geneticists. 1. EXOME SEQUENCING: Exome Sequencing is a very efficient strategy to study most exons of a patient’s genome, unraveling mutations associated with specific disorders or phenotypes. With this diagnostic strategy, patients can be studied with a significantly reduced turnaround time and cost. CGC Genetics has available 2 options for Exome Sequencing: • Whole Exome Sequencing (WES), which analyzes the entire exome (about 20 000 genes); • Disease Exome by CGC Genetics, which analyzes about 6 000 clinically-relevant genes. Any of these can be performed in the index case or in a Trio. 2. NGS PANELS For NGS panels, several genes associated with the same phenotype are simultaneously sequenced. These panels provide increased diagnostic capability with a significantly reduced turnaround time and cost. CGC Genetics has several NGS panels for Ophthalmology that are constantly updated (www.cgcgenetics.com). Any gene studied in exome or NGS panel can also be individually sequenced and analyzed for deletion/duplication events. 3. EXPERTISE IN MEDICAL GENETICS CGC Genetics has Medical Geneticists specialized in genetic counseling for ophthalmological diseases who may advice in choosing the most appropriate -
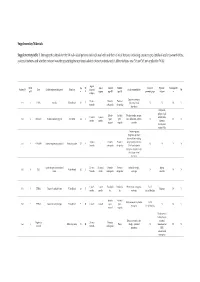
Solved/Unsolved
Supplementary Materials: Supplementary table 1. Demographic details for the 54 individual patients (solved/unsolved) and their clinical features including cataract type, details of ocular co-morbidities, systemic features and whether cataract was the presenting feature (non-isolated cataract patients only). Abbreviations: yes (Y), no (N), not applicable (N/A). Age at Famil Ag M/ Age at Cataract Cataract Cataract Systemic Consanguinit Patient ID Gene Confirmed genetic diagnosis Ethnicity diagnosi Ocular co-morbidities FH y ID e F surgery type RE type LE presenting sign features y s (days) Aniridia, nystagmus, 23 years Posterior Posterior 1-1 1 PAX6 Aniridia White British 25 F - glaucoma, foveal N N N Y 4 months subcapsular subcapsular hypoplasia Cleft palate, epilepsy, high Aphakia Aphakia Macular atrophy, myopia, 7 years 9 7 years 8 arched palate, 2-1 2 COL11A1 Stickler syndrome, type II Not Stated 34 F (post- (post- lens subluxation, vitreous N N N months months flattened surgical) surgical) anomaly maxilla, short stature (5'2ft) Anterior segment dysgenesis, pupillary abnormalities including 12 years Posterior Posterior ectopic pupils, ectropion 3-1 3 CPAMD8 Anterior segment dysgenesis 8 Other, Any other 27 F - N N Y N 5 months subcapsular subcapsular UVAE and irodensis, nystagmus, dysplastic optic discs, large corneal diameters Gyrate atrophy of choroid and 23 years 29 years 1 Posterior Posterior Retinal dystrophy, Bipolar 4-1 4 OAT White British 42 F N N N retina 7 months month subcapsular subcapsular exotropia disorder 1 year 6 1 year -

Feasibility and Outcome of Descemet Membrane Endothelial Keratoplasty in Complex Anterior Segment and Vitreous Disease
CLINICAL SCIENCE Feasibility and Outcome of Descemet Membrane Endothelial Keratoplasty in Complex Anterior Segment and Vitreous Disease Julia M. Weller, MD, Theofilos Tourtas, MD, and Friedrich E. Kruse, MD escemet membrane endothelial keratoplasty (DMEK), Purpose: Descemet membrane endothelial keratoplasty (DMEK) is Da technique for posterior lamellar keratoplasty, involves becoming the method of choice for treating Fuchs endothelial a graft consisting only of the thin Descemet membrane with dystrophy and pseudophakic bullous keratopathy. We investigated adherent corneal endothelial cells. Introduced in 2006 by whether DMEK can serve as a routine procedure in endothelial Melles et al,1 DMEK is becoming more popular as several decompensation even in complex preoperative situations. studies show its superiority to Descemet stripping automated Methods: Of a total of 1184 DMEK surgeries, 24 consecutive eyes endothelial keratoplasty (DSAEK), regarding visual function 2,3 with endothelial decompensation and complex preoperative situa- and the time of visual rehabilitation after DMEK. However, tions were retrospectively analyzed and divided into 5 groups: group because DMEK grafts are thinner than DSAEK grafts, it is fi 1: irido-corneo-endothelial syndrome (n = 3), group 2: aphakia, more dif cult to handle them and typically takes surgeons subluxated posterior chamber intraocular lens or anterior chamber longer to learn. intraocular lens (n = 6), group 3: DMEK after trabeculectomy (n = In difficult situations, most surgeons prefer DSAEK or 4), group 4: DMEK with simultaneous intravitreal injection (n = 6), penetrating keratoplasty to DMEK because of its possible and group 5: DMEK after vitrectomy (n = 5). Main outcome intraoperative complications. For example, if corneal edema 4 parameters were best-corrected visual acuity, central corneal thick- is advanced, Ham et al recommend performing DSAEK first ness, endothelial cell density, rebubbling rate, and graft failure rate. -

A Case of Hallermann-Streiff Syndrome with Aphakia
Korean Journal of Pediatrics Vol. 51, No. 6, 2008 DOI : 10.3345/kjp.2008.51.6.646 Case report 1) A case of Hallermann-Streiff syndrome with aphakia Myung Chul Lee, M.D., Im Jeong Choi, M.D., and Jin Wha Jung, M.D. Department of Pediatrics, Maryknoll Medical Center, Busan, Korea = Abstract = Hallermann-Streiff syndrome is a rare disease. Approximately 150 cases have been reported, including 6 cases in Korea. The authors experienced a case of Hallermann-Streiff syndrome in a 6-year-old female with aphakia. The syndrome is charac- terized by a bird-like face, dental abnormalities, hypotrichosis, atrophy of the skin, bilateral microphthalmia, and proportionate dwarfism. A brief review of the literature was conducted. (Korean J Pediatr 2008;51 :646-649) Key Words : Hallermann-Streiff syndrome, Aphakia, Bird-like face age of 40 weeks and her mother’s obstetric history revealed Introduction no record of systemic disease or drug administration. Her parents and only brother showed no specific finding. On Hallermann-Streiff Syndrome is a rare genetic disorder admission, she was 5 years and 7 months old and her that is characterized by bird-like face, dental abnormalities, height was 83.2 cm (less than 3rd percentile) while her hypotrichosis, atrophy of skin, congenital cataracts, bilateral body weight was 13 kg (less than 3rd percentile), showing microphthalmia, and proportionate nanism. It was first pub- a growth disorder. She showed a developmental disorder lished by Aubry in 18931),butthecasewasincomplete.This showing unassisted self-ambulation at the age of 4 years. syndrome was first described completely in 1948 by She had a pointed nose and frontal bossing as well as Hallermann2) and then in 1950 by Streiff3). -

Journal of Ophthalmology & Clinical Research
ISSN: 2573-9573 Case Report Journal of Ophthalmology & Clinical Research Bilateral Congenital Ectropion Uveae, Anterior Segment Dysgenesis and Aniridia with Microspherophakic Congenital Cataracts and RubeosisIridis Rao Muhammad Arif Khan* and Ashal Kaiser Pal *Corresponding author Rao Muhammad Arif Khan, MCPS, FCPS, FPO, FACS, Pediatric Ophthalmologist, King Edward Medical University, Al-Awali Street, Taif Road, Makkah, Saudi Arabia, Pediatric Ophthalmologist, King Edward Medical University, Tel: 00966560479694; E-mail: [email protected] Makkah, Saudi Arabia Submitted: 02 Apr 2018; Accepted: 12 Apr 2018; Published: 19 Apr 2018 Abstract In recent times, multiple eye diseases have been seen associated with an increase in the rate of Demodex infestation as a possible cause, but in the particular case of dry eye syndrome in patients treated with platelet-rich plasma, this increase in mite may be relevant to guide a more adequate treatment focusing on the elimination of the mite in conjunction with the recovery of the ocular ecology. The demodex mite is a commensal parasite that lives in hair follicles, sebaceous glands and meibomian, which in a high rate of infestation can generate alterations in the ocular area. Performing an adequate diagnosis for the detection of the mite and treatment for its eradication can be effective for the recovery of the normal physiology of the tear film that constitutes a cause of dry eye. Introduction Congenital ectropion uvea is a rare ocular manifestation of neural crest syndrome [1]. It is a non-progressive anomaly characterized by presence of iris pigment epithelium on anterior surface of iris from the pigment ruff [2]. Congenital glaucoma is its common association [3-8].