Page 1 of 23 RSC Advances
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

OPPORTUNITIES ACROSS TAIWAN a Review of 2019’S Investment Trends Sheds Light on Taiwan’S Six Metros
COLLIERS RADAR COMMERCIAL PROPERTY | RESEARCH | TAIPEI | 8 APRIL 2020 Eilleen Liang Director | Research | Taiwan +886 2 8722 8601 [email protected] OPPORTUNITIES ACROSS TAIWAN A review of 2019’s investment trends sheds light on Taiwan’s six metros. COLLIERS RADAR COMMERCIAL PROPERTY | RESEARCH | TAIPEI | 8 APRIL 2020 Insights & Recommendations Annual land Commercial Although Taiwan’s economy has been sales hit NTD276.5 property affected by the US-China trade war in 2019, it also pushed manufacturing and billion in 2019, transactions totaled technology sectors to relocated partial increasing NTD138.6 billion operations back to Taiwan, increasing the demand for office, industrial office and 49.6% YOY up 70.0% YOY factories. Coupled with the low interest rates, the investment amount hit a record high in 2019. Looking forward in 2020, we Top destination for Most stable city for think uncertainties such as outbreak of COVID-19, US-China tensions, and the land investment- commercial cross-strait relations will likely impact the Taichung City property income: investment momentum especially in H1 2020. > Office: We still think the office sector is Taipei City the best opportunity for investors. Though leasing demand will likely > In 2019, land and commercial property investments grew significantly. The total sales value reached slowdown in H1 2020, with latent NTD415.1 billion (USD13.4 billion), a 56% increase compared to 2018. This is also the record high demand and a lack of supply we expect since Colliers survey started in 2007. rents and vacancy to remain stable. > Taiwan’s six metros are destinations both for investors and developers, with a total commercial > Industrial: We recommend owner- property sales value of NTD129.6 billion (USD4.2 billion), 94% of Taiwan’s total. -

Bulletin FEBRUARY 2013
ISSUE 51- 52 Bulletin FEBRUARY 2013 Kaohsiung Exhibition & Convention Center BANGKOK BEIJING HONG KONG SHANGHAI SINGAPORE TAIWAN CONTENTS AWARDS AND RECOGNITIONS 01 MAA Bulletin Issue 51- 52 February 2013 BIM PROJECT CASE STUDY 12 MAA TAIPEI NEW OFFICE 13 PROJECTS 1ST MAY 2011 TO 29TH 14 FEBRUARY 2012 Founded in 1975, MAA is a leading engineering and consulting service provider in the East and Southeast Asian region with a broad range PROFESSIONAL ACTIVITIES 22 of focus areas including infrastructure, land resources, environment, - PROFESSIONAL ACTIVITIES buildings, and information technology. - PROFESSIONAL AWARDS/HONORS - SEMINARS AND CONFERENCE - TECHNICAL PUBLICATIONS To meet the global needs of both public and private clients, MAA has developed sustainable engineering solutions - ranging from PERSONNEL PROFILES 26 conceptual planning, general consultancy, engineering design to project management. MAA employs 1000 professional individuals with offices in the Greater China Region (Beijing, Hong Kong, Shanghai, Taiwan), Mekong Region (Bangkok), and Southeast Asian Region (Singapore), creating a strong professional network in East/Southeast Asia. MAA’s business philosophy is to provide professional services that will become an asset to our clients with long lasting benefits in this rapidly changing social-economic environment. ASSET represents five key components that underlineMAA ’s principles of professional services: Advanced Technology project Safety client’s Satisfaction ISO 9001 and LAB CERTIFICATIONS Economical Solution Timely -

Website : the Bank Website
Website : http://newmaps.twse.com.tw The Bank Website : http://www.landbank.com.tw Time of Publication : July 2018 Spokesman Name: He,Ying-Ming Title: Executive Vice President Tel: (02)2348-3366 E-Mail: [email protected] First Substitute Spokesman Name: Chu,Yu-Feng Title: Executive Vice President Tel: (02) 2348-3686 E-Mail: [email protected] Second Substitute Spokesman Name: Huang,Cheng-Ching Title: Executive Vice President Tel: (02) 2348-3555 E-Mail: [email protected] Address &Tel of the bank’s head office and Branches(please refer to’’ Directory of Head Office and Branches’’) Credit rating agencies Name: Moody’s Investors Service Address: 24/F One Pacific Place 88 Queensway Admiralty, Hong Kong. Tel: (852)3758-1330 Fax: (852)3758-1631 Web Site: http://www.moodys.com Name: Standard & Poor’s Corp. Address: Unit 6901, level 69, International Commerce Centre 1 Austin Road West Kowloon, Hong Kong Tel: (852)2841-1030 Fax: (852)2537-6005 Web Site: http://www.standardandpoors.com Name: Taiwan Ratings Corporation Address: 49F., No7, Sec.5, Xinyi Rd., Xinyi Dist., Taipei City 11049, Taiwan (R.O.C) Tel: (886)2-8722-5800 Fax: (886)2-8722-5879 Web Site: http://www.taiwanratings.com Stock transfer agency Name: Secretariat land bank of Taiwan Co., Ltd. Address: 3F, No.53, Huaining St. Zhongzheng Dist., Taipei City 10046, Taiwan(R,O,C) Tel: (886)2-2348-3456 Fax: (886)2-2375-7023 Web Site: http://www.landbank.com.tw Certified Publick Accountants of financial statements for the past year Name of attesting CPAs: Gau,Wey-Chuan, Mei,Ynan-Chen Name of Accounting Firm: KPMG Addres: 68F., No.7, Sec.5 ,Xinyi Rd., Xinyi Dist., Taipei City 11049, Taiwan (R.O.C) Tel: (886)2-8101-6666 Fax: (886)2-8101-6667 Web Site: http://www.kpmg.com.tw The Bank’s Website: http://www.landbank.com.tw Website: http://newmaps.twse.com.tw The Bank Website: http://www.landbank.com.tw Time of Publication: July 2018 Land Bank of Taiwan Annual Report 2017 Publisher: Land Bank of Taiwan Co., Ltd. -

Characterization of the Biological Activity of a Potent Small Molecule Hec1 Inhibitor TAI-1
Huang et al. Journal of Experimental & Clinical Cancer Research 2014, 33:6 http://www.jeccr.com/content/33/1/6 RESEARCH Open Access Characterization of the biological activity of a potent small molecule Hec1 inhibitor TAI-1 Lynn YL Huang1*, Ying-Shuan Lee2, Jiann-Jyh Huang2, Chia-chi Chang1, Jia-Ming Chang2, Shih-Hsien Chuang2, Kuo-Jang Kao1, Yung-Jen Tsai2, Pei-Yi Tsai2, Chia-Wei Liu2, Her-Sheng Lin2 and Johnson YN Lau1 Abstract Background: Hec1 (NDC80) is an integral part of the kinetochore and is overexpressed in a variety of human cancers, making it an attractive molecular target for the design of novel anticancer therapeutics. A highly potent first-in-class compound targeting Hec1, TAI-1, was identified and is characterized in this study to determine its potential as an anticancer agent for clinical utility. Methods: The in vitro potency, cancer cell specificity, synergy activity, and markers for response of TAI-1 were evaluated with cell lines. Mechanism of action was confirmed with western blotting and immunofluorescent staining. The in vivo potency of TAI-1 was evaluated in three xenograft models in mice. Preliminary toxicity was evaluated in mice. Specificity to the target was tested with a kinase panel. Cardiac safety was evaluated with hERG assay. Clinical correlation was performed with human gene database. Results: TAI-1 showed strong potency across a broad spectrum of tumor cells. TAI-1 disrupted Hec1-Nek2 protein interaction, led to Nek2 degradation, induced significant chromosomal misalignment in metaphase, and induced apoptotic cell death. TAI-1 was effective orally in in vivo animal models of triple negative breast cancer, colon cancer and liver cancer. -

Taiwan High Speed Rail Corporation
Taiwan High Speed Rail Corporation Financial Statements for the Six Months Ended June 30, 2020 and 2019 and Independent Auditors’ Review Report TAIWAN HIGH SPEED RAIL CORPORATION BALANCE SHEETS (In Thousands of New Taiwan Dollars) June 30, 2020 December 31, 2019 June 30, 2019 (Reviewed) (Audited) (Reviewed) ASSETS Amount % Amount % Amount % CURRENT ASSETS Cash and cash equivalents (Note 6) $ 4,935,761 1 $ 16,271,676 4 $ 1,531,221 - Financial assets at fair value through profit or loss (Note 7) 10,452,039 2 330,443 - 329,546 - Notes and accounts receivable (Note 22) 261,521 - 333,092 - 463,471 - Current tax assets (Note 4) - - 166,783 - 166,783 - Inventories (Note 9) 2,447,056 1 2,056,045 1 2,385,672 1 Other financial assets (Notes 10 and 29) 20,401,275 5 22,207,764 5 21,191,885 5 Other current assets (Notes 14 and 28) 1,009,675 - 1,344,333 - 561,045 - Total current assets 39,507,327 9 42,710,136 10 26,629,623 6 NON-CURRENT ASSETS Property, plant and equipment (Note 11) 109,720 - 125,047 - 90,480 - Right-of-use assets (Note 12) 547,287 - 628,988 - 702,947 - Operating concession assets (Notes 13 and 28) 383,796,986 89 390,113,063 88 395,979,795 92 Computer software, net (Note 13) 61,939 - 54,413 - 47,971 - Deferred tax assets (Note 4) 7,429,020 2 7,316,212 2 7,090,054 2 Other financial assets (Notes 10 and 29) 2,058,101 - 2,102,503 - 2,128,646 - Other non-current assets (Note 14) 7,369 - 9,240 - 13,252 - Total non-current assets 394,010,422 91 400,349,466 90 406,053,145 94 TOTAL $ 433,517,749 100 $ 443,059,602 100 $ 432,682,768 100 LIABILITIES -

1 Introduction 2 Features 3 Application
LOCOSYS Technology Inc. 20F.-13, No.79, Sec. 1, Xintai 5th Rd., Xizhi District, New Taipei City 221, Taiwan ℡ 886-2-8698-3698 886-2-8698-3699 www.locosystech.com/ Product name Description Version HD-1612-BA Datasheet of HD-1612-BA standalone GNSS module 1.0 1 Introduction LOCOSYS HD-1612-BA is a complete standalone GNSS module which uses CEC HED latest HD8020 GNSS chip to integrate with an additional LNA and SAW filter. The module can simultaneously acquire and track multiple satellite constellations that include GPS, BEIDOU, GALILEO, QZSS and SBAS. It features low power and small form factor. Besides, it can provide you with superior sensitivity and performance even in urban canyon and dense foliage environment. 2 Features HED high sensitivity solution Support GPS, BEIDOU, GALILEO and QZSS Capable of SBAS (WAAS, EGNOS, MSAS, GAGAN) Support up to 72-channel GNSS Fast TTFF at low signal level Support 1PPS output Built-in DC/DC converter to save power Built-in additional LNA and SAW filter Embedded an active antenna supervisor SMD type; RoHS compliant ISO/TS 16949 quality control 3 Application Personal positioning and navigation Automotive navigation, model aircraft navigation Marine navigation Page 1/18 © 2016 LOCOSYS Technology Inc. LOCOSYS Technology Inc. 20F.-13, No.79, Sec. 1, Xintai 5th Rd., Xizhi District, New Taipei City 221, Taiwan ℡ 886-2-8698-3698 886-2-8698-3699 www.locosystech.com/ 4 Overview 4.1 Block Diagram 4.2 GNSS Performance Chip HD8020 GPS, GALILEO, QZSS: L1 1575.42MHz, C/A code Frequency BEIDOU: B1 1561.098MHz, C/A code Channels Up to 72 channels Update rate 1Hz default Tracking -160dBm up to -161dBm (with external LNA) Sensitivity Cold start -146.5dBm up to -148dBm (with external LNA) Hot start (Open Sky) < 1s (typical) Acquisition Time Cold Start (Open Sky) 28s (typical) Position Accuracy Autonomous 2.5m CEP Max. -

Exhibitor Profiles
Water for the Future summi t&expo Exhibitor Profiles Public Works Department (PWD), Taipei City Government https://pwd.gov.taipei/ Company Profile ‧PWD was established in 1945. Responsible for major public construction work in Taipei City, including New Construction Office, Hydraulic Engineering Office, Parks & Street Lights Office, Sewerage Systems Office and Geotechnical Engineering Office. Product Description Taipei water environment policy:Sponge City ‧Taipei adopts the sponge city concept as the core of its water environment policy. Gradually transforming Taipei into a safe, sustainable, water-friendly, ecological water-front city. The rapid Cellular-Automaton urban flooding model ‧To reduce flooding disasters, PWD and NTU (National Taiwan University) have executed a scientific research plan together which conducts real-time flood assessment and early warning. Taipei Water Department https://www.water.gov.taipei/ Company Profile ‧Taipei Water Department (TWD) was established in 1907. It has been established for more than 100 years. The service area covers Taipei City, Sanchong, Xindian , Yonghe, Zhonghe districts and 7 Li’s of Xizhi District. Product Description District Smart Metering(DSM) ‧Taipei Water Department (TWD) has divided the water distribution network into 319 DMAs as of 2018, and also built relevant management systems to provide long-term results for analysis of water network efficiency. The revenue water rate has reached up to 90%. Direct Drinking ‧More than 500 potable drinking fountains installed across Taipei area are accessible by citizens for their easy use, and the fountain app is also available for citizens to find the nearby ones to drink. Taipei Water Management Office https://www.wratb.gov.tw/ Company Profile ‧Taipei water source district is located in the southeastern of Taipei Metropolitan. -

Bulletin SEPTEMBER 2011
ISSUE 49- 5 0 Bulletin SEPTEMBER 2011 BANGKOK BEIJING HONG KONG SHANGHAI SINGAPORE TAIWAN CONTENTS AwarDS AND RECOGNITIONS 01 MAA Bulletin - THE 10TH (2010) Public construction Golden Issue 49- 50 September 2011 Award - THE 12TH (2010) national architecture Golden ACHIEVEMENT Award AND TOP NationaL ARCHI- TECTURE Award - THE 11TH (2010) Motc Golden Way Award - The 2011 excellent tunnel enGineerinG Award Founded in 1975, MAA is a leading Asian engineering and consulting - Giant GrouP PharMaceutical caMPus Project service provider in the East and Southeast Asian region focused in the – SURV AND MORPHOSIS ARCHITECTS areas of infrastructure, land resources, environment, buildings, and - DiaMond certification for Green buildinG information technology. MateriaL LABEL BUILDING INFormation MODELING 07 To meet the global needs of both public and private clients, MAA (BIM) has a full range of engineering capabilities to provide clients with sustainable solutions - ranging from conceptual planning, general MAA TATPEI OFFICE MOVING AND 09 consultancy, engineering design to project management. NEW OFFICE ESTABLISHED NOTICE PROJECTS 1ST MAY 2010 to 10 MAA employs 800 professional staff people with offices in the Greater 30TH APRIL 2011 China Region (Beijing, Hong Kong, Shanghai, Taiwan), Mekong Region (Bangkok), and Southeast Asia Region (Singapore), creating PROFESSIONAL ACTIVITIES 20 a close professional network in East/Southeast Asia. - PROFESSIONAL ACTIVITIES - PROFESSIONAL Awards/HONORS MAA’s goal is to establish engineering capability that will meet local - SEMINARS AND CONFERENCE needs. Along with the changes in social-economic environment over - TECHNICAL PUBLications the years, MAA’s business philosophy is to provide professional PERSONNEL PROFILES 27 service that will become an asset to clients with long lasting benefits. -

1 Introduction 2 Features 3 Application
LOCOSYS Technology Inc. 20F.-13, No.79, Sec. 1, Xintai 5th Rd., Xizhi Dist., New Taipei City 221, Taiwan ℡ 886-2-8698-3698 886-2-8698-3699 www.locosystech.com/ Product name Description Version MC-1612-G2 Standalone multiple GNSS module 1.0 1 Introduction LOCOSYS MC-1612-G2 is a complete standalone GNSS module. The module can simultaneously acquire and track multiple satellite constellations that include GPS, GLONASS, GALILEO, QZSS and SBAS. It features low power and small form factor. Besides, it can provide you with superior sensitivity and performance even in urban canyon and dense foliage environment. This module supports hybrid ephemeris prediction to achieve faster cold start. One is self-generated ephemeris prediction (called EASY) that is no need of both network assistance and host CPU’s intervention. This is valid for up to 3 days and updates automatically from time to time when GNSS module is powered on and satellites are available. The other is server-generated ephemeris prediction (called EPO) that gets from an internet server. This is valid for up to 14 days. Both ephemeris predictions are stored in the on-board flash memory and perform a cold start time less than 15 seconds. 2 Features MediaTek high sensitivity solution Support GPS, GLONASS, GALILEO and QZSS Capable of SBAS (WAAS, EGNOS, MSAS, GAGAN) Support 99-channel GNSS Low power consumption Fast TTFF at low signal level Built-in 12 multi-tone active interference canceller Free hybrid ephemeris prediction to achieve faster cold start Built-in data logger Up to 10 Hz update rate ±11ns high accuracy time pulse (1PPS) Indoor and outdoor multi-path detection and compensation Small form factor 16 x 12.2 x 2.2 mm SMD type with stamp holes; RoHS compliant ISO/TS 16949 quality control Support RTCM SC-104 Version-2.x 3 Application Personal positioning and navigation Automotive navigation Page 1/23 © 2014 LOCOSYS Technology Inc. -

Land Development
Land Development •Land Development Analysis •Environment Data Analysis •Development Plan •Engineering Design •Financial Plan •Project & Construction Management MAA Sustainable Engineering Action Land Development MAA Sustainable Eng. Concepts Founded in 1975, MAA is a leading engineering and consulting service provider in the East and Land Development Southeast Asian region with a broad range of focus areas including infrastructure, land resources, environment, buildings, and information technology. To meet the global needs of both public and private clients, MAA has developed sustainable engineering solutions - ranging from conceptual planning, general consultancy, engineering design to project management. • Sustainable City MAA employs 1000 professional individuals • Industrial & Commercial Parks with offices in the Greater China Region (Beijing, • Urban Planning & Land Use Developments Hong Kong, Shanghai, Taiwan), Mekong Region • New Towns & New Communities (Bangkok), and Southeast Asian Region (Singapore), creating a strong professional network in East/ • Sports & Leisure Southeast Asia. Create a Green MAA’s business philosophy is to provide Environment professional services that will become an asset to our clients with long lasting benefits in this rapidly changing social-economic environment. ASSET represents five key components that underline MAA’s principles of professional services: Advanced technology Adoption of Green Engineering Methods project Safety client’s Satisfaction Economical solution Timely completion Selection of -
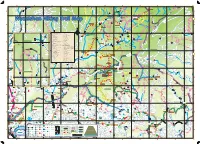
Wuzeishan Hiking Trail Map Lane 95 Bridge Connecting Point to the Small Picture on the Left
(Lane 43, Pingjing St, Industry Rd) Pinding Ancient Canal Ancient Pinding L a n S27 e Mt. Xinzhentou Dingshan 9 3 , (Xiajingque Industry Rd) Mt. Xingdui P i n g j i n Jingque Stream Jingque g Taozijiao Bridge S t ( N e Fengguizui Trail i l Mt. Neishuanxi i a Ciyun o nt C e I a (Xiaguoxi Industry Rd) Industry (Xiaguoxi Temple n ci nal d u s Stamp Pavilion An Wuzhishan Industry Rd t r Hi y g Qingfeng Pavilion n Tianweizi Bridge Wanxi Industry Rd ) d R i k d i g ng n Xiwan Rd Pi T r Pingjing St ail (Chencuo Industry Rd) S19 Neicuo Neicuo Bridge Shilin District, Taipei Sec 3, Zhishan Rd Xizhi District, New Taipei Daqitou Trail Chedengjiao Wuzeishan Hiking Trail Map Lane 95 Bridge Connecting point to the small picture on the left. Connecting point to the small picture ry Rd) Mt. Ewei ndust S I h u a St, n g g x i n g o ji Mt. Shuanxi u Mt. Wuzhishan g H i s in t P o r , i c Pingjing St (Pingxi Industry Rd) Industry (Pingxi St Pingjing Pingdeng S18 T 106 Shengren r (Lane 42, Pingjing St, Industry Rd) a Zhulin Trail Pingdingguchaun i Wanxi Industry Rd l (Lane Hiking Trail Entrance Waterfall N e Mt. Suiwei i Lane 371 s h (Lane 105, Pingjing St, Industry Rd) Industry St, Pingjing 105, (Lane u a n S Pingxi Trail t r e Mt. Meihua a Zhengtian Gongping Bridge m Temple Xiwan 3rd Bridge D6 Nature Trail Index of the Trails in Taipei S18 D7 The North Trail Shengren E5 Daluntoushan and Dalunweishan Shilin District, Taipei Xiwan Rd Yongzhen Trail Bridge Shengren Shanxi Touristic TaiPower Electricity Hiking Trail E6 Yuanming Temple Trail Bridge Farm Relay Station E5 Zhongshe Rd Branch Guandi Lutian Xinyi Rd B7 Daqitou Trail Temple Huaxin 2nd Rd E6 Bishan Rd Branch Mt. -

Taiwan High Speed Rail Corporation
Taiwan High Speed Rail Corporation Financial Statements for the Nine Months Ended September 30, 2020 and 2019 and Independent Auditors’ Review Report TAIWAN HIGH SPEED RAIL CORPORATION BALANCE SHEETS (In Thousands of New Taiwan Dollars) September 30, 2020 December 31, 2019 September 30, 2019 (Reviewed) (Audited) (Reviewed) ASSETS Amount % Amount % Amount % CURRENT ASSETS Cash and cash equivalents (Note 6) $ 1,283,279 - $ 16,271,676 4 $ 769,619 - Financial assets at fair value through profit or loss (Note 7) 13,178,348 3 330,443 - 330,005 - Notes and accounts receivable (Note 22) 279,079 - 333,092 - 569,047 - Current tax assets (Note 4) - - 166,783 - 166,783 - Inventories (Note 9) 2,217,239 1 2,056,045 1 2,305,846 1 Other financial assets (Notes 10 and 29) 14,314,130 4 22,207,764 5 21,938,840 5 Other current assets (Notes 14 and 28) 801,869 - 1,344,333 - 709,662 - Total current assets 32,073,944 8 42,710,136 10 26,789,802 6 NON-CURRENT ASSETS Property, plant and equipment (Note 11) 102,424 - 125,047 - 84,771 - Right-of-use assets (Note 12) 506,723 - 628,988 - 665,436 - Operating concession assets (Notes 13 and 28) 380,808,033 90 390,113,063 88 392,808,580 91 Computer software, net (Note 13) 82,066 - 54,413 - 44,781 - Deferred tax assets (Note 4) 7,512,003 2 7,316,212 2 7,218,013 2 Other financial assets (Notes 10 and 29) 2,056,334 - 2,102,503 - 2,128,646 1 Other non-current assets (Note 14) 6,243 - 9,240 - 11,205 - Total non-current assets 391,073,826 92 400,349,466 90 402,961,432 94 TOTAL $ 423,147,770 100 $ 443,059,602 100 $ 429,751,234