Parallel Framework for Inferring Genome Scale Gene Regulatory Networks
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Downloaded the Normalized Gene Expression Profile from the GEO (Package Geoquery (5))
1 SUPPLEMENTARY MATERIALS 2 Supplementary Methods 3 Study design 4 The workflow to identify and validate the TME risk score and TME subtypes in gastric cancer is 5 depicted in Fig. S1. We first estimated the absolute abundance levels of the major stromal and 6 immune cell types in the TME using bulk gene expression data, and assessed the prognostic 7 effect of these cells in a discovery cohort. Next, we constructed a TME risk score and validated it 8 in two independent gene expression validation cohorts and three immunohistochemistry 9 validation cohorts. Finally, we stratified patients into four TME subtypes and examined their 10 genomic and molecular features and relation to established molecular subtypes. 11 Gene expression data 12 To explore the prognostic landscape of the TME, we used four gene expression profile (GEP) 13 datasets of resected gastric cancer patients with publicly available clinical information, namely, 14 ACRG (GSE62254) (1), GSE15459 (2), GSE84437 (3), and TCGA stomach adenocarcinoma 15 (STAD). Specifically, the raw microarray data in the ACRG cohort and GSE15459 were retrieved 16 from the Gene Expression Omnibus (GEO), and normalized by the RMA algorithm (package affy) 17 using custom chip definition files (Brainarray version 23 (4)) that convert Affymetrix probesets to 18 Entrez gene IDs. For GSE84437 dataset, which was measured by the Illumina platform, we 19 downloaded the normalized gene expression profile from the GEO (package GEOquery (5)). The 20 Illumina probes were also mapped to Entrez genes. For multiple probes mapping to the same 21 Entrez gene, we selected the one with the maximum mean expression level as the surrogate for 22 the Entrez gene using the function of collapseRows (6) (package WGCNA). -

Mouse Steap4 Conditional Knockout Project (CRISPR/Cas9)
https://www.alphaknockout.com Mouse Steap4 Conditional Knockout Project (CRISPR/Cas9) Objective: To create a Steap4 conditional knockout Mouse model (C57BL/6J) by CRISPR/Cas-mediated genome engineering. Strategy summary: The Steap4 gene (NCBI Reference Sequence: NM_054098 ; Ensembl: ENSMUSG00000012428 ) is located on Mouse chromosome 5. 5 exons are identified, with the ATG start codon in exon 2 and the TAA stop codon in exon 5 (Transcript: ENSMUST00000115421). Exon 2~3 will be selected as conditional knockout region (cKO region). Deletion of this region should result in the loss of function of the Mouse Steap4 gene. To engineer the targeting vector, homologous arms and cKO region will be generated by PCR using BAC clone RP23-212F6 as template. Cas9, gRNA and targeting vector will be co-injected into fertilized eggs for cKO Mouse production. The pups will be genotyped by PCR followed by sequencing analysis. Note: Mice homozygous for a knock-out allele exhibit adipose accumulation, oxidative stress, increased liver weight, lower metabolic rate, hypoactivity, insulin resistance, glucose intolerance, mild hyperglycemia and dyslipidemia. Exon 2 starts from about 100% of the coding region. The knockout of Exon 2~3 will result in frameshift of the gene. The size of intron 1 for 5'-loxP site insertion: 14897 bp, and the size of intron 3 for 3'-loxP site insertion: 1385 bp. The size of effective cKO region: ~2082 bp. The cKO region does not have any other known gene. Page 1 of 7 https://www.alphaknockout.com Overview of the Targeting Strategy Wildtype allele 5' gRNA region gRNA region 3' 1 2 3 4 5 Targeting vector Targeted allele Constitutive KO allele (After Cre recombination) Legends Exon of mouse Steap4 Homology arm cKO region loxP site Page 2 of 7 https://www.alphaknockout.com Overview of the Dot Plot Window size: 10 bp Forward Reverse Complement Sequence 12 Note: The sequence of homologous arms and cKO region is aligned with itself to determine if there are tandem repeats. -

Analysis of the Indacaterol-Regulated Transcriptome in Human Airway
Supplemental material to this article can be found at: http://jpet.aspetjournals.org/content/suppl/2018/04/13/jpet.118.249292.DC1 1521-0103/366/1/220–236$35.00 https://doi.org/10.1124/jpet.118.249292 THE JOURNAL OF PHARMACOLOGY AND EXPERIMENTAL THERAPEUTICS J Pharmacol Exp Ther 366:220–236, July 2018 Copyright ª 2018 by The American Society for Pharmacology and Experimental Therapeutics Analysis of the Indacaterol-Regulated Transcriptome in Human Airway Epithelial Cells Implicates Gene Expression Changes in the s Adverse and Therapeutic Effects of b2-Adrenoceptor Agonists Dong Yan, Omar Hamed, Taruna Joshi,1 Mahmoud M. Mostafa, Kyla C. Jamieson, Radhika Joshi, Robert Newton, and Mark A. Giembycz Departments of Physiology and Pharmacology (D.Y., O.H., T.J., K.C.J., R.J., M.A.G.) and Cell Biology and Anatomy (M.M.M., R.N.), Snyder Institute for Chronic Diseases, Cumming School of Medicine, University of Calgary, Calgary, Alberta, Canada Received March 22, 2018; accepted April 11, 2018 Downloaded from ABSTRACT The contribution of gene expression changes to the adverse and activity, and positive regulation of neutrophil chemotaxis. The therapeutic effects of b2-adrenoceptor agonists in asthma was general enriched GO term extracellular space was also associ- investigated using human airway epithelial cells as a therapeu- ated with indacaterol-induced genes, and many of those, in- tically relevant target. Operational model-fitting established that cluding CRISPLD2, DMBT1, GAS1, and SOCS3, have putative jpet.aspetjournals.org the long-acting b2-adrenoceptor agonists (LABA) indacaterol, anti-inflammatory, antibacterial, and/or antiviral activity. Numer- salmeterol, formoterol, and picumeterol were full agonists on ous indacaterol-regulated genes were also induced or repressed BEAS-2B cells transfected with a cAMP-response element in BEAS-2B cells and human primary bronchial epithelial cells by reporter but differed in efficacy (indacaterol $ formoterol . -

Single-Cell Analysis Uncovers Fibroblast Heterogeneity
ARTICLE https://doi.org/10.1038/s41467-020-17740-1 OPEN Single-cell analysis uncovers fibroblast heterogeneity and criteria for fibroblast and mural cell identification and discrimination ✉ Lars Muhl 1,2 , Guillem Genové 1,2, Stefanos Leptidis 1,2, Jianping Liu 1,2, Liqun He3,4, Giuseppe Mocci1,2, Ying Sun4, Sonja Gustafsson1,2, Byambajav Buyandelger1,2, Indira V. Chivukula1,2, Åsa Segerstolpe1,2,5, Elisabeth Raschperger1,2, Emil M. Hansson1,2, Johan L. M. Björkegren 1,2,6, Xiao-Rong Peng7, ✉ Michael Vanlandewijck1,2,4, Urban Lendahl1,8 & Christer Betsholtz 1,2,4 1234567890():,; Many important cell types in adult vertebrates have a mesenchymal origin, including fibro- blasts and vascular mural cells. Although their biological importance is undisputed, the level of mesenchymal cell heterogeneity within and between organs, while appreciated, has not been analyzed in detail. Here, we compare single-cell transcriptional profiles of fibroblasts and vascular mural cells across four murine muscular organs: heart, skeletal muscle, intestine and bladder. We reveal gene expression signatures that demarcate fibroblasts from mural cells and provide molecular signatures for cell subtype identification. We observe striking inter- and intra-organ heterogeneity amongst the fibroblasts, primarily reflecting differences in the expression of extracellular matrix components. Fibroblast subtypes localize to discrete anatomical positions offering novel predictions about physiological function(s) and regulatory signaling circuits. Our data shed new light on the diversity of poorly defined classes of cells and provide a foundation for improved understanding of their roles in physiological and pathological processes. 1 Karolinska Institutet/AstraZeneca Integrated Cardio Metabolic Centre, Blickagången 6, SE-14157 Huddinge, Sweden. -
Primepcr™Assay Validation Report
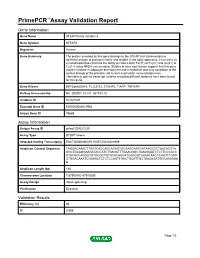
PrimePCR™Assay Validation Report Gene Information Gene Name STEAP family member 4 Gene Symbol STEAP4 Organism Human Gene Summary The protein encoded by this gene belongs to the STEAP (six transmembrane epithelial antigen of prostate) family and resides in the golgi apparatus. It functions as a metalloreductase that has the ability to reduce both Fe(3+) to Fe(2+) and Cu(2+) to Cu(1+) using NAD(+) as acceptor. Studies in mice and human suggest that this gene maybe involved in adipocyte development and metabolism and may contribute to the normal biology of the prostate cell as well as prostate cancer progression. Alternatively spliced transcript variants encoding different isoforms have been found for this gene. Gene Aliases DKFZp666D049, FLJ23153, STAMP2, TIARP, TNFAIP9 RefSeq Accession No. NC_000007.13, NT_007933.15 UniGene ID Hs.521008 Ensembl Gene ID ENSG00000127954 Entrez Gene ID 79689 Assay Information Unique Assay ID qHsaCID0021230 Assay Type SYBR® Green Detected Coding Transcript(s) ENST00000380079, ENST00000301959 Amplicon Context Sequence TAGGACAAACTTGATCACCAGCACAGTGCAAGGAATGATAAGCCCTAACACGTA GGCTGCAGGAAGATACCATCTGAGATTTGAAGGGCTGAGGAATCTCTTCCCACC GTACACCAGGGTGTGGGCTGTACACAAGATCAGGGTCAAATAACCCAGTTTGGA CTGGACAAATCGGAACTCTCTCCAGTTGACTGCATTGCTAACAGATGGCAAAGAA G Amplicon Length (bp) 188 Chromosome Location 7:87908782-87910285 Assay Design Intron-spanning Purification Desalted Validation Results Efficiency (%) 96 R2 0.999 Page 1/5 PrimePCR™Assay Validation Report cDNA Cq 22.3 cDNA Tm (Celsius) 84 gDNA Cq 38.74 Specificity (%) 100 Information -

ID AKI Vs Control Fold Change P Value Symbol Entrez Gene Name *In
ID AKI vs control P value Symbol Entrez Gene Name *In case of multiple probesets per gene, one with the highest fold change was selected. Fold Change 208083_s_at 7.88 0.000932 ITGB6 integrin, beta 6 202376_at 6.12 0.000518 SERPINA3 serpin peptidase inhibitor, clade A (alpha-1 antiproteinase, antitrypsin), member 3 1553575_at 5.62 0.0033 MT-ND6 NADH dehydrogenase, subunit 6 (complex I) 212768_s_at 5.50 0.000896 OLFM4 olfactomedin 4 206157_at 5.26 0.00177 PTX3 pentraxin 3, long 212531_at 4.26 0.00405 LCN2 lipocalin 2 215646_s_at 4.13 0.00408 VCAN versican 202018_s_at 4.12 0.0318 LTF lactotransferrin 203021_at 4.05 0.0129 SLPI secretory leukocyte peptidase inhibitor 222486_s_at 4.03 0.000329 ADAMTS1 ADAM metallopeptidase with thrombospondin type 1 motif, 1 1552439_s_at 3.82 0.000714 MEGF11 multiple EGF-like-domains 11 210602_s_at 3.74 0.000408 CDH6 cadherin 6, type 2, K-cadherin (fetal kidney) 229947_at 3.62 0.00843 PI15 peptidase inhibitor 15 204006_s_at 3.39 0.00241 FCGR3A Fc fragment of IgG, low affinity IIIa, receptor (CD16a) 202238_s_at 3.29 0.00492 NNMT nicotinamide N-methyltransferase 202917_s_at 3.20 0.00369 S100A8 S100 calcium binding protein A8 215223_s_at 3.17 0.000516 SOD2 superoxide dismutase 2, mitochondrial 204627_s_at 3.04 0.00619 ITGB3 integrin, beta 3 (platelet glycoprotein IIIa, antigen CD61) 223217_s_at 2.99 0.00397 NFKBIZ nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitor, zeta 231067_s_at 2.97 0.00681 AKAP12 A kinase (PRKA) anchor protein 12 224917_at 2.94 0.00256 VMP1/ mir-21likely ortholog -

Single-Cell RNA Sequencing of Human, Macaque, and Mouse Testes Uncovers Conserved and Divergent Features of Mammalian Spermatogenesis
bioRxiv preprint doi: https://doi.org/10.1101/2020.03.17.994509; this version posted March 18, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Single-cell RNA sequencing of human, macaque, and mouse testes uncovers conserved and divergent features of mammalian spermatogenesis Adrienne Niederriter Shami1,7, Xianing Zheng1,7, Sarah K. Munyoki2,7, Qianyi Ma1, Gabriel L. Manske3, Christopher D. Green1, Meena Sukhwani2, , Kyle E. Orwig2*, Jun Z. Li1,4*, Saher Sue Hammoud1,3,5,6,8* 1Department of Human Genetics, University of Michigan, Ann Arbor, MI, USA 2 Department of Obstetrics, Gynecology and Reproductive Sciences, Integrative Systems Biology Graduate Program, Magee-Womens Research Institute, University of Pittsburgh School of Medicine, Pittsburgh, PA. 3 Cellular and Molecular Biology Program, University of Michigan, Ann Arbor, MI, USA 4 Department of Computational Medicine and Bioinformatics, University of Michigan, Ann Arbor, MI, USA 5 Department of Obstetrics and Gynecology, University of Michigan, Ann Arbor, MI, USA 6Department of Urology, University of Michigan, Ann Arbor, MI, USA 7 These authors contributed equally 8 Lead Contact * Correspondence: [email protected] (K.E.O.), [email protected] (J.Z.L.), [email protected] (S.S.H.) bioRxiv preprint doi: https://doi.org/10.1101/2020.03.17.994509; this version posted March 18, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Summary Spermatogenesis is a highly regulated process that produces sperm to transmit genetic information to the next generation. -

Entrez ID Gene Name Fold Change Q-Value Description
Entrez ID gene name fold change q-value description 4283 CXCL9 -7.25 5.28E-05 chemokine (C-X-C motif) ligand 9 3627 CXCL10 -6.88 6.58E-05 chemokine (C-X-C motif) ligand 10 6373 CXCL11 -5.65 3.69E-04 chemokine (C-X-C motif) ligand 11 405753 DUOXA2 -3.97 3.05E-06 dual oxidase maturation factor 2 4843 NOS2 -3.62 5.43E-03 nitric oxide synthase 2, inducible 50506 DUOX2 -3.24 5.01E-06 dual oxidase 2 6355 CCL8 -3.07 3.67E-03 chemokine (C-C motif) ligand 8 10964 IFI44L -3.06 4.43E-04 interferon-induced protein 44-like 115362 GBP5 -2.94 6.83E-04 guanylate binding protein 5 3620 IDO1 -2.91 5.65E-06 indoleamine 2,3-dioxygenase 1 8519 IFITM1 -2.67 5.65E-06 interferon induced transmembrane protein 1 3433 IFIT2 -2.61 2.28E-03 interferon-induced protein with tetratricopeptide repeats 2 54898 ELOVL2 -2.61 4.38E-07 ELOVL fatty acid elongase 2 2892 GRIA3 -2.60 3.06E-05 glutamate receptor, ionotropic, AMPA 3 6376 CX3CL1 -2.57 4.43E-04 chemokine (C-X3-C motif) ligand 1 7098 TLR3 -2.55 5.76E-06 toll-like receptor 3 79689 STEAP4 -2.50 8.35E-05 STEAP family member 4 3434 IFIT1 -2.48 2.64E-03 interferon-induced protein with tetratricopeptide repeats 1 4321 MMP12 -2.45 2.30E-04 matrix metallopeptidase 12 (macrophage elastase) 10826 FAXDC2 -2.42 5.01E-06 fatty acid hydroxylase domain containing 2 8626 TP63 -2.41 2.02E-05 tumor protein p63 64577 ALDH8A1 -2.41 6.05E-06 aldehyde dehydrogenase 8 family, member A1 8740 TNFSF14 -2.40 6.35E-05 tumor necrosis factor (ligand) superfamily, member 14 10417 SPON2 -2.39 2.46E-06 spondin 2, extracellular matrix protein 3437 -

Transcriptome Analysis of Traf6 Function in the Innate Immune
Innate immune signalling of the zebrafish embryo Stockhammer, O.W. Citation Stockhammer, O. W. (2010, May 19). Innate immune signalling of the zebrafish embryo. Retrieved from https://hdl.handle.net/1887/15512 Version: Corrected Publisher’s Version Licence agreement concerning inclusion of doctoral License: thesis in the Institutional Repository of the University of Leiden Downloaded from: https://hdl.handle.net/1887/15512 Note: To cite this publication please use the final published version (if applicable). Transcriptome analysis of Traf6 function in the 4 innate immune response of zebrafish embryos Oliver W. Stockhammer1, Han Rauwerda2, Floyd R. Wittink2, Timo M. Breit2, Annemarie H. Meijer1 and Herman P. Spaink1 1 Institute of Biology, Leiden University, Leiden, The Netherlands 2 MicroArray Department & Integrative Bioinformatics Unit, Swammerdam Institute for Life Sciences, University of Amsterdam, Amsterdam, The Netherlands Chapter 4 Abstract TRAF6 is a key player at the cross-roads of development and immunity. The analy- sis of its in vivo molecular function is a great challenge since severe developmental defects and early lethality caused by Traf6 deficiency in knock-out mice interfere with analyses of the immune response. In this study we have used a new strategy to analyse the function of Traf6 in a zebrafish-Salmonella infectious disease model. In our approach the effect of a Traf6 translation-blocking morpholino was titrated down to avoid developmental defects and the response to infection under these par- tial knock-down conditions was studied using the combination of microarray and next generation sequencing technology. Transcriptome profiling of the traf6 knock- down allowed the identification of a gene set whose responsiveness during infec- tion is highly dependent on Traf6. -

Content Based Search in Gene Expression Databases and a Meta-Analysis of Host Responses to Infection
Content Based Search in Gene Expression Databases and a Meta-analysis of Host Responses to Infection A Thesis Submitted to the Faculty of Drexel University by Francis X. Bell in partial fulfillment of the requirements for the degree of Doctor of Philosophy November 2015 c Copyright 2015 Francis X. Bell. All Rights Reserved. ii Acknowledgments I would like to acknowledge and thank my advisor, Dr. Ahmet Sacan. Without his advice, support, and patience I would not have been able to accomplish all that I have. I would also like to thank my committee members and the Biomed Faculty that have guided me. I would like to give a special thanks for the members of the bioinformatics lab, in particular the members of the Sacan lab: Rehman Qureshi, Daisy Heng Yang, April Chunyu Zhao, and Yiqian Zhou. Thank you for creating a pleasant and friendly environment in the lab. I give the members of my family my sincerest gratitude for all that they have done for me. I cannot begin to repay my parents for their sacrifices. I am eternally grateful for everything they have done. The support of my sisters and their encouragement gave me the strength to persevere to the end. iii Table of Contents LIST OF TABLES.......................................................................... vii LIST OF FIGURES ........................................................................ xiv ABSTRACT ................................................................................ xvii 1. A BRIEF INTRODUCTION TO GENE EXPRESSION............................. 1 1.1 Central Dogma of Molecular Biology........................................... 1 1.1.1 Basic Transfers .......................................................... 1 1.1.2 Uncommon Transfers ................................................... 3 1.2 Gene Expression ................................................................. 4 1.2.1 Estimating Gene Expression ............................................ 4 1.2.2 DNA Microarrays ...................................................... -

Deep Sequencing of the Zebrafish Transcriptome Response to Mycobacterium Infection
Transcriptome profiling of infectious diseases and cancer in zebrafish Ordas, A.K. Citation Ordas, A. K. (2010, June 29). Transcriptome profiling of infectious diseases and cancer in zebrafish. Retrieved from https://hdl.handle.net/1887/15734 Version: Not Applicable (or Unknown) License: Leiden University Non-exclusive license Downloaded from: https://hdl.handle.net/1887/15734 Note: To cite this publication please use the final published version (if applicable). Chapter 2 Deep sequencing of the zebrafish transcriptome response to mycobacterium infection This chapter is based on: Hegedus Z, Zakrzewska A, Agoston VC, Ordas A, Racz P, Mink M, Spaink HP, Meijer AH. Deep sequencing of the zebrafish transcriptome response to myco- bacterium infection. Mol Immunol. 2009 Sep;46(15):2918-30. Chapter 2 Abstract Novel high-throughput deep sequencing technology has dramatically changed the way that the functional complexity of transcriptomes can be studied. Here we report on the first use of this technology to gain insight into the wide range of transcrip- tional responses that are associated with an infectious disease process. Using Solexa/ Illumina’s digital gene expression (DGE) system, a tag-based transcriptome sequenc- ing method, we investigated mycobacterium-induced transcriptome changes in a model vertebrate species, the zebrafish. We obtained a sequencing depth of over 5 million tags per sample with strong correlation between replicates. Tag mapping indicated that healthy and infected adult zebrafish express over 70% of all genes represented in transcript databases. Comparison of the data with a previous mul- ti-platform microarray analysis showed that both types of technologies identified regulation of similar functional groups of genes. -

Jimmunol.1800667.Full.Pdf
Placenta Specific 8 Suppresses IL-18 Production through Regulation of Autophagy and Is Associated with Adult Still Disease This information is current as Seiji Segawa, Yuya Kondo, Yuji Nakai, Akira Iizuka, of September 25, 2021. Shunta Kaneko, Masahiro Yokosawa, Kotona Furuyama, Hiroto Tsuboi, Daisuke Goto, Isao Matsumoto and Takayuki Sumida J Immunol published online 7 November 2018 http://www.jimmunol.org/content/early/2018/11/06/jimmun Downloaded from ol.1800667 Supplementary http://www.jimmunol.org/content/suppl/2018/11/06/jimmunol.180066 Material 7.DCSupplemental http://www.jimmunol.org/ Why The JI? Submit online. • Rapid Reviews! 30 days* from submission to initial decision • No Triage! Every submission reviewed by practicing scientists by guest on September 25, 2021 • Fast Publication! 4 weeks from acceptance to publication *average Subscription Information about subscribing to The Journal of Immunology is online at: http://jimmunol.org/subscription Permissions Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Email Alerts Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts The Journal of Immunology is published twice each month by The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2018 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. Published November 7, 2018, doi:10.4049/jimmunol.1800667 The Journal of Immunology Placenta Specific 8 Suppresses IL-18 Production through Regulation of Autophagy and Is Associated with Adult Still Disease Seiji Segawa,* Yuya Kondo,* Yuji Nakai,† Akira Iizuka,* Shunta Kaneko,* Masahiro Yokosawa,* Kotona Furuyama,* Hiroto Tsuboi,* Daisuke Goto,* Isao Matsumoto,* and Takayuki Sumida* Adult Still disease (ASD) is a systemic disorder of unknown etiology characterized by high spiking fever, rash, and arthritis.