Keratinocyte-Specific Deletion of SHARPIN Induces Atopic Dermatitis-Like Inflammation in Mice
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Small Cell Ovarian Carcinoma: Genomic Stability and Responsiveness to Therapeutics
Gamwell et al. Orphanet Journal of Rare Diseases 2013, 8:33 http://www.ojrd.com/content/8/1/33 RESEARCH Open Access Small cell ovarian carcinoma: genomic stability and responsiveness to therapeutics Lisa F Gamwell1,2, Karen Gambaro3, Maria Merziotis2, Colleen Crane2, Suzanna L Arcand4, Valerie Bourada1,2, Christopher Davis2, Jeremy A Squire6, David G Huntsman7,8, Patricia N Tonin3,4,5 and Barbara C Vanderhyden1,2* Abstract Background: The biology of small cell ovarian carcinoma of the hypercalcemic type (SCCOHT), which is a rare and aggressive form of ovarian cancer, is poorly understood. Tumourigenicity, in vitro growth characteristics, genetic and genomic anomalies, and sensitivity to standard and novel chemotherapeutic treatments were investigated in the unique SCCOHT cell line, BIN-67, to provide further insight in the biology of this rare type of ovarian cancer. Method: The tumourigenic potential of BIN-67 cells was determined and the tumours formed in a xenograft model was compared to human SCCOHT. DNA sequencing, spectral karyotyping and high density SNP array analysis was performed. The sensitivity of the BIN-67 cells to standard chemotherapeutic agents and to vesicular stomatitis virus (VSV) and the JX-594 vaccinia virus was tested. Results: BIN-67 cells were capable of forming spheroids in hanging drop cultures. When xenografted into immunodeficient mice, BIN-67 cells developed into tumours that reflected the hypercalcemia and histology of human SCCOHT, notably intense expression of WT-1 and vimentin, and lack of expression of inhibin. Somatic mutations in TP53 and the most common activating mutations in KRAS and BRAF were not found in BIN-67 cells by DNA sequencing. -
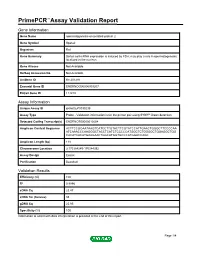
Primepcr™Assay Validation Report
PrimePCR™Assay Validation Report Gene Information Gene Name spermatogenesis-associated protein 2 Gene Symbol Spata2 Organism Rat Gene Summary Sertoli cell mRNA expression is induced by FSH; may play a role in spermatogenesis; localized to the nucleus Gene Aliases Not Available RefSeq Accession No. Not Available UniGene ID Rn.201291 Ensembl Gene ID ENSRNOG00000009207 Entrez Gene ID 114210 Assay Information Unique Assay ID qRnoCEP0030238 Assay Type Probe - Validation information is for the primer pair using SYBR® Green detection Detected Coding Transcript(s) ENSRNOT00000012604 Amplicon Context Sequence ACTTCCGGAATAAGTCATCCTTGTACTTCGTATCCATTGAACTGGGCTTCCCCAA ATCAAACCCAAGGGCTACCTCATCTCCCCCATGGCTCTGGGGCTGGAGGCTGG CACATCACATGAAGAACTGGCATGGTGCCCATGGACCAGC Amplicon Length (bp) 118 Chromosome Location 3:170384245-170384392 Assay Design Exonic Purification Desalted Validation Results Efficiency (%) 100 R2 0.9996 cDNA Cq 23.47 cDNA Tm (Celsius) 85 gDNA Cq 25.95 Specificity (%) 100 Information to assist with data interpretation is provided at the end of this report. Page 1/4 PrimePCR™Assay Validation Report Spata2, Rat Amplification Plot Amplification of cDNA generated from 25 ng of universal reference RNA Melt Peak Melt curve analysis of above amplification Standard Curve Standard curve generated using 20 million copies of template diluted 10-fold to 20 copies Page 2/4 PrimePCR™Assay Validation Report Products used to generate validation data Real-Time PCR Instrument CFX384 Real-Time PCR Detection System Reverse Transcription Reagent iScript™ Advanced cDNA Synthesis Kit for RT-qPCR Real-Time PCR Supermix SsoAdvanced™ SYBR® Green Supermix Experimental Sample qPCR Reference Total RNA Data Interpretation Unique Assay ID This is a unique identifier that can be used to identify the assay in the literature and online. Detected Coding Transcript(s) This is a list of the Ensembl transcript ID(s) that this assay will detect. -

Analysis of Gene Expression Data for Gene Ontology
ANALYSIS OF GENE EXPRESSION DATA FOR GENE ONTOLOGY BASED PROTEIN FUNCTION PREDICTION A Thesis Presented to The Graduate Faculty of The University of Akron In Partial Fulfillment of the Requirements for the Degree Master of Science Robert Daniel Macholan May 2011 ANALYSIS OF GENE EXPRESSION DATA FOR GENE ONTOLOGY BASED PROTEIN FUNCTION PREDICTION Robert Daniel Macholan Thesis Approved: Accepted: _______________________________ _______________________________ Advisor Department Chair Dr. Zhong-Hui Duan Dr. Chien-Chung Chan _______________________________ _______________________________ Committee Member Dean of the College Dr. Chien-Chung Chan Dr. Chand K. Midha _______________________________ _______________________________ Committee Member Dean of the Graduate School Dr. Yingcai Xiao Dr. George R. Newkome _______________________________ Date ii ABSTRACT A tremendous increase in genomic data has encouraged biologists to turn to bioinformatics in order to assist in its interpretation and processing. One of the present challenges that need to be overcome in order to understand this data more completely is the development of a reliable method to accurately predict the function of a protein from its genomic information. This study focuses on developing an effective algorithm for protein function prediction. The algorithm is based on proteins that have similar expression patterns. The similarity of the expression data is determined using a novel measure, the slope matrix. The slope matrix introduces a normalized method for the comparison of expression levels throughout a proteome. The algorithm is tested using real microarray gene expression data. Their functions are characterized using gene ontology annotations. The results of the case study indicate the protein function prediction algorithm developed is comparable to the prediction algorithms that are based on the annotations of homologous proteins. -

Molecular and Physiological Basis for Hair Loss in Near Naked Hairless and Oak Ridge Rhino-Like Mouse Models: Tracking the Role of the Hairless Gene
University of Tennessee, Knoxville TRACE: Tennessee Research and Creative Exchange Doctoral Dissertations Graduate School 5-2006 Molecular and Physiological Basis for Hair Loss in Near Naked Hairless and Oak Ridge Rhino-like Mouse Models: Tracking the Role of the Hairless Gene Yutao Liu University of Tennessee - Knoxville Follow this and additional works at: https://trace.tennessee.edu/utk_graddiss Part of the Life Sciences Commons Recommended Citation Liu, Yutao, "Molecular and Physiological Basis for Hair Loss in Near Naked Hairless and Oak Ridge Rhino- like Mouse Models: Tracking the Role of the Hairless Gene. " PhD diss., University of Tennessee, 2006. https://trace.tennessee.edu/utk_graddiss/1824 This Dissertation is brought to you for free and open access by the Graduate School at TRACE: Tennessee Research and Creative Exchange. It has been accepted for inclusion in Doctoral Dissertations by an authorized administrator of TRACE: Tennessee Research and Creative Exchange. For more information, please contact [email protected]. To the Graduate Council: I am submitting herewith a dissertation written by Yutao Liu entitled "Molecular and Physiological Basis for Hair Loss in Near Naked Hairless and Oak Ridge Rhino-like Mouse Models: Tracking the Role of the Hairless Gene." I have examined the final electronic copy of this dissertation for form and content and recommend that it be accepted in partial fulfillment of the requirements for the degree of Doctor of Philosophy, with a major in Life Sciences. Brynn H. Voy, Major Professor We have read this dissertation and recommend its acceptance: Naima Moustaid-Moussa, Yisong Wang, Rogert Hettich Accepted for the Council: Carolyn R. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Genetics of Amyotrophic Lateral Sclerosis in the Han Chinese
Genetics of amyotrophic lateral sclerosis in the Han Chinese Ji He A thesis submitted for the degree of Master of Philosophy at The University of Queensland in 2015 The University of Queensland Diamantina Institute 1 Abstract Amyotrophic lateral sclerosis is the most frequently occurring neuromuscular degenerative disorders, and has an obscure aetiology. Whilst major progress has been made, the majority of the genetic variation involved in ALS is, as yet, undefined. In this thesis, multiple genetic studies have been conducted to advance our understanding of the genetic architecture of the disease. In the light of the paucity of comprehensive genetic studies performed in Chinese, the presented study focused on advancing our current understanding in genetics of ALS in the Han Chinese population. To identify genetic variants altering risk of ALS, a genome-wide association study (GWAS) was performed. The study included 1,324 Chinese ALS cases and 3,115 controls. After quality control, a number of analyses were performed in a cleaned dataset of 1,243 cases and 2,854 controls that included: a genome-wide association analysis to identify SNPs associated with ALS; a genomic restricted maximum likelihood (GREML) analysis to estimate the proportion of the phenotypic variance in ALS liability due to common SNPs; and a gene- based analysis to identify genes associated with ALS. There were no genome-wide significant SNPs or genes associated with ALS. However, it was estimated that 17% (SE: 0.05; P=6×10-5) of the phenotypic variance in ALS liability was due to common SNPs. The top associated SNP was within GNAS (rs4812037; p =7×10-7). -

Human S100A4 (P9ka) Induces the Metastatic Phenotype Upon Benign Tumour Cells
Oncogene (1998) 17, 465 ± 473 1998 Stockton Press All rights reserved 0950 ± 9232/98 $12.00 http://www.stockton-press.co.uk/onc Human S100A4 (p9Ka) induces the metastatic phenotype upon benign tumour cells Bryony H Lloyd, Angela Platt-Higgins, Philip S Rudland and Roger Barraclough School of Biological Sciences, University of Liverpool, PO Box 147, Liverpool, L69 7ZB, UK The rodent S100-related calcium-binding protein, strengthened by the observation that transgenic mice, S100A4 induces metastasis in non-metastatic rat and expressing elevated levels of S100A4 (p9Ka) from 17 mouse benign mammary cells and co-operates with additional, position-independent rat S100A4 trans- benign-tumour-inducing changes in two transgenic mouse genes, fail to show any detectable phenotype (Davies, models, to yield metastatic mammary tumours. Co- 1993). When these mice are mated with transgenic mice transfection of the human gene for S100A4 with containing additional copies of the activated rodent c- pSV2neo into the benign rat mammary cell line, Rama erbB-2 oncogene, neu, under the control of the MMTV 37, yielded cells which expressed a low level of the LTR (Bouchard et al., 1989), ospring inheriting the endogenous S100A4 mRNA, and either high or neu oncogene suer sporadic mammary tumours after undetectable levels of human S100A4 mRNA. The cells 12 ± 14 months of repeated mating. In contrast which expressed a high level of human S100A4 mRNA ospring inheriting both neu and S100A4 transgenes induced metastasis in the benign rat mammary cell line show an elevated incidence of primary tumours, and, in Rama 37 in an in vivo assay, whereas the cells which addition, metastases in the lungs which account for up expressed an undetectable level of human S100A4 did to 24% of the total lung area (Davies et al., 1996). -

4-6 Weeks Old Female C57BL/6 Mice Obtained from Jackson Labs Were Used for Cell Isolation
Methods Mice: 4-6 weeks old female C57BL/6 mice obtained from Jackson labs were used for cell isolation. Female Foxp3-IRES-GFP reporter mice (1), backcrossed to B6/C57 background for 10 generations, were used for the isolation of naïve CD4 and naïve CD8 cells for the RNAseq experiments. The mice were housed in pathogen-free animal facility in the La Jolla Institute for Allergy and Immunology and were used according to protocols approved by the Institutional Animal Care and use Committee. Preparation of cells: Subsets of thymocytes were isolated by cell sorting as previously described (2), after cell surface staining using CD4 (GK1.5), CD8 (53-6.7), CD3ε (145- 2C11), CD24 (M1/69) (all from Biolegend). DP cells: CD4+CD8 int/hi; CD4 SP cells: CD4CD3 hi, CD24 int/lo; CD8 SP cells: CD8 int/hi CD4 CD3 hi, CD24 int/lo (Fig S2). Peripheral subsets were isolated after pooling spleen and lymph nodes. T cells were enriched by negative isolation using Dynabeads (Dynabeads untouched mouse T cells, 11413D, Invitrogen). After surface staining for CD4 (GK1.5), CD8 (53-6.7), CD62L (MEL-14), CD25 (PC61) and CD44 (IM7), naïve CD4+CD62L hiCD25-CD44lo and naïve CD8+CD62L hiCD25-CD44lo were obtained by sorting (BD FACS Aria). Additionally, for the RNAseq experiments, CD4 and CD8 naïve cells were isolated by sorting T cells from the Foxp3- IRES-GFP mice: CD4+CD62LhiCD25–CD44lo GFP(FOXP3)– and CD8+CD62LhiCD25– CD44lo GFP(FOXP3)– (antibodies were from Biolegend). In some cases, naïve CD4 cells were cultured in vitro under Th1 or Th2 polarizing conditions (3, 4). -

The Capacity of Long-Term in Vitro Proliferation of Acute Myeloid
The Capacity of Long-Term in Vitro Proliferation of Acute Myeloid Leukemia Cells Supported Only by Exogenous Cytokines Is Associated with a Patient Subset with Adverse Outcome Annette K. Brenner, Elise Aasebø, Maria Hernandez-Valladares, Frode Selheim, Frode Berven, Ida-Sofie Grønningsæter, Sushma Bartaula-Brevik and Øystein Bruserud Supplementary Material S2 of S31 Table S1. Detailed information about the 68 AML patients included in the study. # of blasts Viability Proliferation Cytokine Viable cells Change in ID Gender Age Etiology FAB Cytogenetics Mutations CD34 Colonies (109/L) (%) 48 h (cpm) secretion (106) 5 weeks phenotype 1 M 42 de novo 241 M2 normal Flt3 pos 31.0 3848 low 0.24 7 yes 2 M 82 MF 12.4 M2 t(9;22) wt pos 81.6 74,686 low 1.43 969 yes 3 F 49 CML/relapse 149 M2 complex n.d. pos 26.2 3472 low 0.08 n.d. no 4 M 33 de novo 62.0 M2 normal wt pos 67.5 6206 low 0.08 6.5 no 5 M 71 relapse 91.0 M4 normal NPM1 pos 63.5 21,331 low 0.17 n.d. yes 6 M 83 de novo 109 M1 n.d. wt pos 19.1 8764 low 1.65 693 no 7 F 77 MDS 26.4 M1 normal wt pos 89.4 53,799 high 3.43 2746 no 8 M 46 de novo 26.9 M1 normal NPM1 n.d. n.d. 3472 low 1.56 n.d. no 9 M 68 MF 50.8 M4 normal D835 pos 69.4 1640 low 0.08 n.d. -

Genome-Wide Sirna Screen for Mediators of NF-Κb Activation
Genome-wide siRNA screen for mediators SEE COMMENTARY of NF-κB activation Benjamin E. Gewurza, Fadi Towficb,c,1, Jessica C. Marb,d,1, Nicholas P. Shinnersa,1, Kaoru Takasakia, Bo Zhaoa, Ellen D. Cahir-McFarlanda, John Quackenbushe, Ramnik J. Xavierb,c, and Elliott Kieffa,2 aDepartment of Medicine and Microbiology and Molecular Genetics, Channing Laboratory, Brigham and Women’s Hospital and Harvard Medical School, Boston, MA 02115; bCenter for Computational and Integrative Biology, Massachusetts General Hospital, Harvard Medical School, Boston, MA 02114; cProgram in Medical and Population Genetics, The Broad Institute of Massachusetts Institute of Technology and Harvard, Cambridge, MA 02142; dDepartment of Biostatistics, Harvard School of Public Health, Boston, MA 02115; and eDepartment of Biostatistics and Computational Biology and Department of Cancer Biology, Dana-Farber Cancer Institute, Boston, MA 02115 Contributed by Elliott Kieff, December 16, 2011 (sent for review October 2, 2011) Although canonical NFκB is frequently critical for cell proliferation, (RIPK1). TRADD engages TNFR-associated factor 2 (TRAF2), survival, or differentiation, NFκB hyperactivation can cause malig- which recruits the ubiquitin (Ub) E2 ligase UBC5 and the E3 nant, inflammatory, or autoimmune disorders. Despite intensive ligases cIAP1 and cIAP2. CIAP1/2 polyubiquitinate RIPK1 and study, mammalian NFκB pathway loss-of-function RNAi analyses TRAF2, which recruit and activate the K63-Ub binding proteins have been limited to specific protein classes. We therefore under- TAB1, TAB2, and TAB3, as well as their associated kinase took a human genome-wide siRNA screen for novel NFκB activa- MAP3K7 (TAK1). TAK1 in turn phosphorylates IKKβ activa- tion pathway components. Using an Epstein Barr virus latent tion loop serines to promote IKK activity (4). -

Noelia Díaz Blanco
Effects of environmental factors on the gonadal transcriptome of European sea bass (Dicentrarchus labrax), juvenile growth and sex ratios Noelia Díaz Blanco Ph.D. thesis 2014 Submitted in partial fulfillment of the requirements for the Ph.D. degree from the Universitat Pompeu Fabra (UPF). This work has been carried out at the Group of Biology of Reproduction (GBR), at the Department of Renewable Marine Resources of the Institute of Marine Sciences (ICM-CSIC). Thesis supervisor: Dr. Francesc Piferrer Professor d’Investigació Institut de Ciències del Mar (ICM-CSIC) i ii A mis padres A Xavi iii iv Acknowledgements This thesis has been made possible by the support of many people who in one way or another, many times unknowingly, gave me the strength to overcome this "long and winding road". First of all, I would like to thank my supervisor, Dr. Francesc Piferrer, for his patience, guidance and wise advice throughout all this Ph.D. experience. But above all, for the trust he placed on me almost seven years ago when he offered me the opportunity to be part of his team. Thanks also for teaching me how to question always everything, for sharing with me your enthusiasm for science and for giving me the opportunity of learning from you by participating in many projects, collaborations and scientific meetings. I am also thankful to my colleagues (former and present Group of Biology of Reproduction members) for your support and encouragement throughout this journey. To the “exGBRs”, thanks for helping me with my first steps into this world. Working as an undergrad with you Dr. -

Absence of S100A4 in the Mouse Lens Induces an Aberrant Retina-Specific Differentiation Program and Cataract
www.nature.com/scientificreports OPEN Absence of S100A4 in the mouse lens induces an aberrant retina‑specifc diferentiation program and cataract Rupalatha Maddala1*, Junyuan Gao2, Richard T. Mathias2, Tylor R. Lewis1, Vadim Y. Arshavsky1,3, Adriana Levine4, Jonathan M. Backer4,5, Anne R. Bresnick4 & Ponugoti V. Rao1,3* S100A4, a member of the S100 family of multifunctional calcium‑binding proteins, participates in several physiological and pathological processes. In this study, we demonstrate that S100A4 expression is robustly induced in diferentiating fber cells of the ocular lens and that S100A4 (−/−) knockout mice develop late‑onset cortical cataracts. Transcriptome profling of lenses from S100A4 (−/−) mice revealed a robust increase in the expression of multiple photoreceptor‑ and Müller glia‑specifc genes, as well as the olfactory sensory neuron‑specifc gene, S100A5. This aberrant transcriptional profle is characterized by corresponding increases in the levels of proteins encoded by the aberrantly upregulated genes. Ingenuity pathway network and curated pathway analyses of diferentially expressed genes in S100A4 (−/−) lenses identifed Crx and Nrl transcription factors as the most signifcant upstream regulators, and revealed that many of the upregulated genes possess promoters containing a high‑density of CpG islands bearing trimethylation marks at histone H3K27 and/or H3K4, respectively. In support of this fnding, we further documented that S100A4 (−/−) knockout lenses have altered levels of trimethylated H3K27 and H3K4. Taken together,