The Evolution, Diversity, and Host Associations of Rhabdoviruses Ben Longdon,1,* Gemma G
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Evolution, Diversity, and Host Associations of Rhabdoviruses Ben Longdon,1,* Gemma G
Virus Evolution, 2015, 1(1): vev014 doi: 10.1093/ve/vev014 Research article The evolution, diversity, and host associations of rhabdoviruses Ben Longdon,1,* Gemma G. R. Murray,1 William J. Palmer,1 Jonathan P. Day,1 Darren J Parker,2,3 John J. Welch,1 Darren J. Obbard4 and Francis M. Jiggins1 1 2 Department of Genetics, University of Cambridge, Cambridge, CB2 3EH, School of Biology, University of Downloaded from St Andrews, St Andrews, KY19 9ST, UK, 3Department of Biological and Environmental Science, University of Jyva¨skyla¨, Jyva¨skyla¨, Finland and 4Institute of Evolutionary Biology, and Centre for Immunity Infection and Evolution, University of Edinburgh, Edinburgh, EH9 3JT, UK *Corresponding author: E-mail: [email protected] http://ve.oxfordjournals.org/ Abstract Metagenomic studies are leading to the discovery of a hidden diversity of RNA viruses. These new viruses are poorly characterized and new approaches are needed predict the host species these viruses pose a risk to. The rhabdoviruses are a diverse family of RNA viruses that includes important pathogens of humans, animals, and plants. We have discovered thirty-two new rhabdoviruses through a combination of our own RNA sequencing of insects and searching public sequence databases. Combining these with previously known sequences we reconstructed the phylogeny of 195 rhabdovirus by guest on December 14, 2015 sequences, and produced the most in depth analysis of the family to date. In most cases we know nothing about the biology of the viruses beyond the host they were identified from, but our dataset provides a powerful phylogenetic approach to predict which are vector-borne viruses and which are specific to vertebrates or arthropods. -

The Evolution, Diversity and Host Associations of Rhabdoviruses
Edinburgh Research Explorer The evolution, diversity and host associations of rhabdoviruses Citation for published version: Obbard, D, Longdon, B, Murray, GGR, Palmer, WJ, Day, JP, Parker, DJ, Welch, JJ & Jiggins, F 2015, 'The evolution, diversity and host associations of rhabdoviruses', Virus Evolution, vol. 1, no. 1, vev014. https://doi.org/10.1093/ve/vev014 Digital Object Identifier (DOI): 10.1093/ve/vev014 Link: Link to publication record in Edinburgh Research Explorer Document Version: Publisher's PDF, also known as Version of record Published In: Virus Evolution General rights Copyright for the publications made accessible via the Edinburgh Research Explorer is retained by the author(s) and / or other copyright owners and it is a condition of accessing these publications that users recognise and abide by the legal requirements associated with these rights. Take down policy The University of Edinburgh has made every reasonable effort to ensure that Edinburgh Research Explorer content complies with UK legislation. If you believe that the public display of this file breaches copyright please contact [email protected] providing details, and we will remove access to the work immediately and investigate your claim. Download date: 30. Sep. 2021 Virus Evolution, 2015, 1(1): vev014 doi: 10.1093/ve/vev014 Research article The evolution, diversity, and host associations of rhabdoviruses Ben Longdon,1,* Gemma G. R. Murray,1 William J. Palmer,1 Jonathan P. Day,1 Darren J Parker,2,3 John J. Welch,1 Darren J. Obbard4 and Francis M. Jiggins1 -

2011 Biodiversity Snapshot. Isle of Man Appendices
UK Overseas Territories and Crown Dependencies: 2011 Biodiversity snapshot. Isle of Man: Appendices. Author: Elizabeth Charter Principal Biodiversity Officer (Strategy and Advocacy). Department of Environment, Food and Agriculture, Isle of man. More information available at: www.gov.im/defa/ This section includes a series of appendices that provide additional information relating to that provided in the Isle of Man chapter of the publication: UK Overseas Territories and Crown Dependencies: 2011 Biodiversity snapshot. All information relating to the Isle or Man is available at http://jncc.defra.gov.uk/page-5819 The entire publication is available for download at http://jncc.defra.gov.uk/page-5821 1 Table of Contents Appendix 1: Multilateral Environmental Agreements ..................................................................... 3 Appendix 2 National Wildife Legislation ......................................................................................... 5 Appendix 3: Protected Areas .......................................................................................................... 6 Appendix 4: Institutional Arrangements ........................................................................................ 10 Appendix 5: Research priorities .................................................................................................... 13 Appendix 6 Ecosystem/habitats ................................................................................................... 14 Appendix 7: Species .................................................................................................................... -

In Istria in Autumn
Go Slow…. In Istria in Autumn Naturetrek Tour Report 1 - 8 October 2019 Althea cannabina Flame Brocade Spiranthes spiralis Monkodonja Report and images by Paul Tout & Paul Harmes Naturetrek Mingledown Barn Wolf's Lane Chawton Alton Hampshire GU34 3HJ UK T: +44 (0)1962 733051 E: [email protected] W: www.naturetrek.co.uk Tour Report Go Slow…. In Istria in Autumn Tour participants: Paul Tout and Paul Harmes (leaders) with 14 Naturetrek clients Day 1 Tuesday 1st October Trieste Airport - Istarske Toplice (our hotel accommodation) Paul Harmes met up with the group at Stansted. Arriving at Trieste airport just after 5pm, we passed through passport control, collected our luggage and moved out into the Arrivals hall where we were met by Paul Tout. We were soon on our way towards Istria, passing through the very attractive historic centre of Trieste and on towards Koper-Capodistria, the main port in Slovenia. At the crossroads of Europe where the three main language groups meet (Romance, Slav and Germanic), the area is an ethnic mix with large areas of bi- (and even tri-) lingualism, so place names are usually hyphenated, made up of two languages. The weather was fine, sunny and warm. We took the road for the centre of Trieste along the Costiera. but no Alpine Swifts were visible. They nest in a colony on the cliffs beside the road and they continue to visit the colonies until mid-October but evidently, they were feeding high up in the fine weather. Passing through the centre of Trieste, the city looked very fine in the evening light, with its Viennese-style waterfront and main square, Piazza Unita d’Italia, the only one in Italy that opens onto the sea. -

Scottish Macro-Moth List, 2015
Notes on the Scottish Macro-moth List, 2015 This list aims to include every species of macro-moth reliably recorded in Scotland, with an assessment of its Scottish status, as guidance for observers contributing to the National Moth Recording Scheme (NMRS). It updates and amends the previous lists of 2009, 2011, 2012 & 2014. The requirement for inclusion on this checklist is a minimum of one record that is beyond reasonable doubt. Plausible but unproven species are relegated to an appendix, awaiting confirmation or further records. Unlikely species and known errors are omitted altogether, even if published records exist. Note that inclusion in the Scottish Invertebrate Records Index (SIRI) does not imply credibility. At one time or another, virtually every macro-moth on the British list has been reported from Scotland. Many of these claims are almost certainly misidentifications or other errors, including name confusion. However, because the County Moth Recorder (CMR) has the final say, dubious Scottish records for some unlikely species appear in the NMRS dataset. A modern complication involves the unwitting transportation of moths inside the traps of visiting lepidopterists. Then on the first night of their stay they record a species never seen before or afterwards by the local observers. Various such instances are known or suspected, including three for my own vice-county of Banffshire. Surprising species found in visitors’ traps the first time they are used here should always be regarded with caution. Clerical slips – the wrong scientific name scribbled in a notebook – have long caused confusion. An even greater modern problem involves errors when computerising the data. -

East Devon Pebblebed Heaths Providing Space for Nature Biodiversity Audit 2016 Space for Nature Report: East Devon Pebblebed Heaths
East Devon Pebblebed Heaths East Devon Pebblebed Providing Space for East Devon Nature Pebblebed Heaths Providing Space for Nature Dr. Samuel G. M. Bridgewater and Lesley M. Kerry Biodiversity Audit 2016 Site of Special Scientific Interest Special Area of Conservation Special Protection Area Biodiversity Audit 2016 Space for Nature Report: East Devon Pebblebed Heaths Contents Introduction by 22nd Baron Clinton . 4 Methodology . 23 Designations . 24 Acknowledgements . 6 European Legislation and European Protected Species and Habitats. 25 Summary . 7 Species of Principal Importance and Introduction . 11 Biodiversity Action Plan Priority Species . 25 Geology . 13 Birds of Conservation Concern . 26 Biodiversity studies . 13 Endangered, Nationally Notable and Nationally Scarce Species . 26 Vegetation . 13 The Nature of Devon: A Biodiversity Birds . 13 and Geodiversity Action Plan . 26 Mammals . 14 Reptiles . 14 Results and Discussion . 27 Butterflies. 14 Species diversity . 28 Odonata . 14 Heathland versus non-heathland specialists . 30 Other Invertebrates . 15 Conservation Designations . 31 Conservation Status . 15 Ecosystem Services . 31 Ownership of ‘the Commons’ and management . 16 Future Priorities . 32 Cultural Significance . 16 Vegetation and Plant Life . 33 Recreation . 16 Existing Condition of the SSSI . 35 Military training . 17 Brief characterisation of the vegetation Archaeology . 17 communities . 37 Threats . 18 The flora of the Pebblebed Heaths . 38 Military and recreational pressure . 18 Plants of conservation significance . 38 Climate Change . 18 Invasive Plants . 41 Acid and nitrogen deposition. 18 Funding and Management Change . 19 Appendix 1. List of Vascular Plant Species . 42 Management . 19 Appendix 2. List of Ferns, Horsetails and Clubmosses . 58 Scrub Clearance . 20 Grazing . 20 Appendix 3. List of Bryophytes . 58 Mowing and Flailing . -

Introduction to Macro-Moth Identification
Introduction to Macro-moth Identification David Slade Lepidoptera County Recorder (VC41) Introduction to Macro-moth Identification • What are moths? • How do you identify them • Moth families • Mysteries and pitfalls What is a Moth? Wikipedia says… The moths are a paraphyletic group of insects related to the butterflies and belonging to the order Lepidoptera. Most lepidopterans are moths and there are thought to be approximately 160,000 species of moth, many of which are yet to be described. Most species of moth are nocturnal, but there are also crepuscular and diurnal species. a group is said to be paraphyletic if it consists of all the descendants of the last common ancestor of the group's members minus a small number of monophyletic groups of descendants, typically just one or two such group Wikipedia also says… Lepidoptera is a large order of insects…. It is one of the most widespread and widely recognizable insect orders in the world. The term was coined by Linnaeus in 1735 and is derived from Ancient Greek λεπίδος (scale) and πτερόν (wing). Lepidoptera show many variations of the basic body structure that have evolved to gain advantages in lifestyle and distribution. It is among the four most speciose orders, along with the Hymenoptera, Diptera, and the Coleoptera.[1] Lepidopteran species are characterized by scales covering their bodies and wings, and a proboscis. The scales are modified, flattened "hairs", and give butterflies and moths their extraordinary variety of colours and patterns. Almost all species have some form of membranous wings, except for a few that have reduced wings or are wingless. -

The Evolution, Diversity and Host Associations of Rhabdoviruses
bioRxiv preprint doi: https://doi.org/10.1101/020107; this version posted September 25, 2015. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC 4.0 International license. 1 1 The evolution, diversity and host associations of rhabdoviruses 2 3 Ben Longdon1*, Gemma GR Murray1, William J Palmer1, Jonathan P Day1, Darren J 4 Parker2, 3, John J Welch1, Darren J Obbard4 and Francis M Jiggins1. 5 6 1Department of Genetics 7 University of Cambridge 8 Cambridge 9 CB2 3EH 10 UK 11 12 2 School of Biology 13 University of St. Andrews 14 St. Andrews 15 KY19 9ST 16 UK 17 18 3Department of Biological and Environmental Science, 19 University of Jyväskylä, 20 Jyväskylä, 21 Finland 22 23 4Institute of Evolutionary Biology, and Centre for Immunity Infection and Evolution 24 University of Edinburgh 25 Edinburgh 26 EH9 3JT 27 UK 28 29 *corresponding author 30 email: [email protected] 31 phone: +441223333945 32 33 bioRxiv preprint doi: https://doi.org/10.1101/020107; this version posted September 25, 2015. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC 4.0 International license. 2 34 Abstract 35 36 Metagenomic studies are leading to the discovery of a hidden diversity of RNA viruses. -

2013 RNHS Annual Report
RNHS 48th Annual Report 2013 The RUTLAND NATURAL HISTORY SOCIETY was founded in February 1965 and now has over 300 members. The objectives of the Society are to further the cause of Natural History, to study, record and help preserve Rutland’s wildlife in particular, to meet and exchange information with other bodies, and to encourage people to take an interest in wildlife and its conservation. The Society organises monthly field outings to places of interest both inside and outside the County, holds indoor meetings on the first Tuesday of each month between October and April, publishes a newsletter, Fieldfare, six times a year, and an Annual Report, and maintains a Website (www.rnhs.org.uk). NB Sightings are documented; Recorders or Secretary may be contacted for details. Contents Officers, Committee and Recorders 2 Chairman’s Report 3 Treasurer’s Report 5 Weather Report 7 Amphibian and Reptile Report 9 Bird Report 10 Wildfowl and Wader counts 18 Botany Report: Verges 23 Roadside verge map 27 Entomology 28 Lepidoptera: butterflies 28 Lepidoptera: moths 31 Orthoptera Report 40 Plant Gall Report 43 Insects and others 46 Glow-worm Report 49 Mammal Report 51 Bat Report 55 RNHS indoor and field meetings 60 Items for loan to RNHS members 68 List of Recorders and contributors inside front cover Site abbreviations inside back cover Cover drawing of Apion frumentarium, by Jacqueline Wright, Shotover Wildlife, Oxford. County Recorder of Bryophytes for Oxfordshire and Berkshire. (The 2 mm long Apion was drawn from a pinned specimen looking down the microscope with the pencil (5H) work done under a magnifying lens by the side of the microscope.) Printed by Lonsdale Printing, Wellingborough 1 RNHS 48th Annual Report 2013 Rutland Natural History Society www.rnhs.org.uk President E. -

Species Recorded Within and Around the Village of Kirkgunzeon
Species recorded within and around the village of Kirkgunzeon The following represents a list of the species recorded within 1km of the village of Kirkgunzeon (NX8666). The list was produced by South West Scotland Environmental Information Centre (SWSEIC) in Feb 2021. Taxon Group Recommended Common Name Recommended Name acarine (Acari) Aceria nalepai Aceria nalepai acarine (Acari) Eriophyes laevis Eriophyes laevis bird Mallard Anas platyrhynchos bird Greylag Goose Anser anser bird Pink-footed Goose Anser brachyrhynchus bird Shelduck Tadorna tadorna bird Oystercatcher Haematopus ostralegus bird European Herring Gull Larus argentatus bird Lesser Black-backed Gull Larus fuscus bird Curlew Numenius arquata bird Grey Heron Ardea cinerea bird Little Egret Egretta garzetta bird Woodpigeon Columba palumbus bird Collared Dove Streptopelia decaocto bird Kingfisher Alcedo atthis bird Cuckoo Cuculus canorus bird Sparrowhawk Accipiter nisus bird Buzzard Buteo buteo bird Red Kite Milvus milvus bird Peregrine Falco peregrinus bird Kestrel Falco tinnunculus bird Red-legged Partridge Alectoris rufa bird Moorhen Gallinula chloropus bird Eurasian Skylark Alauda arvensis bird Treecreeper Certhia familiaris bird Dipper Cinclus cinclus bird Northern Raven Corvus corax bird Carrion Crow Corvus corone bird Rook Corvus frugilegus bird Eurasian Magpie Pica pica bird Yellowhammer Emberiza citrinella bird Common Reed Bunting Emberiza schoeniclus bird Goldfinch Carduelis carduelis bird Greenfinch Chloris chloris bird Common Chaffinch Fringilla coelebs bird Eurasian -
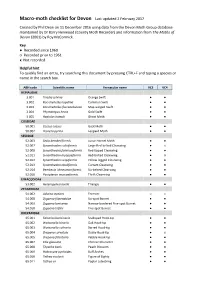
Macro-Moth Checklist for Devon Last Updated 2 February 2017
Macro-moth checklist for Devon Last updated 2 February 2017 Created by Phil Dean on 11 December 2016 using data from the Devon Moth Group database maintained by Dr Barry Henwood (County Moth Recorder) and information from The Moths of Devon (2001) by Roy McCormick. Key ● Recorded since 1960 ○ Recorded prior to 1961 x Not recorded Helpful hint To quickly find an entry, try searching this document by pressing CTRL+F and typing a species or name in the search box. ABH code Scientific name Vernacular name VC3 VC4 HEPIALIDAE 3.001 Triodia sylvina Orange Swift ● ● 3.002 Korscheltellus lupulina Common Swift ● ● 3.003 Korscheltellus fusconebulosa Map-winged Swift ● ● 3.004 Phymatopus hecta Gold Swift ● ● 3.005 Hepialus humuli Ghost Moth ● ● COSSIDAE 50.001 Cossus cossus Goat Moth ● ● 50.002 Zeuzera pyrina Leopard Moth ● ● SESIIDAE 52.003 Sesia bembeciformis Lunar Hornet Moth ● ● 52.007 Synanthedon culiciformis Large Red-belted Clearwing ● x 52.008 Synanthedon formicaeformis Red-tipped Clearwing ● ● 52.011 Synanthedon myopaeformis Red-belted Clearwing ● ○ 52.012 Synanthedon vespiformis Yellow-legged Clearwing ● ● 52.013 Synanthedon tipuliformis Currant Clearwing ● ● 52.014 Bembecia ichneumoniformis Six-belted Clearwing ● ● 52.016 Pyropteron muscaeformis Thrift Clearwing ● ● LIMACODIDAE 53.002 Heterogenea asella Triangle ● ● ZYGAENIDAE 54.002 Adscita statices Forester ○ x 54.008 Zygaena filipendulae Six-spot Burnet ● ● 54.009 Zygaena lonicerae Narrow-bordered Five-spot Burnet ● ● 54.010 Zygaena trifolii Five-spot Burnet ● ● DREPANIDAE 65.001 Falcaria -
Montrose Basin Lepidoptera Records Page 1
Montrose Basin Lepidoptera Records 0 9 C V Code Taxon Vernacular s Grid ref. / sq. & Site Last Record Larval Foodplants u t a t S 3.001 Hepialus sylvina Orange Swift 2 NO 691565 MBVC 23.07.09 Polyphagous (low plants) - roots 3.002 Hepialus lupulinus Common Swift 2 NO 691565 MBVC 14.06.11 Roots of Grasses 3.003 K.fusconebulosa Map-winged Swift 2 NO 701565 MBVC 13.07.15 Bracken roots 3.005 Hepialus humuli Ghost Moth 1 NO 691565 MBVC 14.06.11 Grasses & other low plants - roots 12.032 Tinea semifulvella NO 701565 MBVC 13.07.15 Wool, Clothes and Brids Nests 12.033 Tinea trinotella NO 701565 MBVC 30.06.15 Birds Nests 16.001 Yponomeuta evonymella Bird-cherry Ermine NO 701565 MBVC 28.08.15 Bird Cherry 16.014 Pseudoswammerdamia combinella NO 691565 MBVC 14.06.11 Blackthorn 16.017 Swammerdamia pyrella NO 691565 MBVC 08.07.11 Hawthorn, Apple, Pear 17.003 Ypsolopha dentella Honeysuckle Moth NO 691565 MBVC 26.08.11 Honeysuckle 17.011 Ypsolopha ustella NO 67-57- Lurgies 04.03.09 Oak 17.012 Ypsolopha sequella NO 701565 MBVC 08.10.15 Sycamore & Field Maple 18.001 Plutella xylostella Diamond-backed Moth NO 701565 MBVC 14.04.15 Brassicas 19.007 Glyphipterix simpliciella Cocksfoot Moth NO 675592 B of D Rly. 22.05.12 Cocksfoot Grass 20.011 Argyresthia brockeella NO 701565 MBVC 03.07.14 Birch or Alder catkins 20.012 Argyresthia goedartella NO 701565 MBVC 28.08.15 Birch or Alder Catkins 20.022 Argyresthia bonnetella NO 701565 MBVC 10.08.15 Hawthorn 22.002 Prays fraxinella Ash Bud Moth NO 691565 MBVC 16.07.12 Ash 28.009 Endrosis sarcitrella White-shouldered House-moth NO 702565 MBRes 09.08.13 Dried plant & animal debris 28.010 H.pseudospretella Brown House-moth NO 701565 MBVC 03.07.14 Dead animal & vegetable matter 28.019 Esperia sulphurella NO 691565 MBVC 17.03.10 Rotten wood 29.001 Diurnea fagella NO 701565 MBVC 14.04.15 Deciduous trees 32.017 Agonopterix arenella NO 701565 MBVC 07.04.15 Thistles & Knapweed 32.018 Agonopterix heracliana .