Genetic–Epigenetic Interactions in Cis: a Major Focus In
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Macropinocytosis Requires Gal-3 in a Subset of Patient-Derived Glioblastoma Stem Cells
ARTICLE https://doi.org/10.1038/s42003-021-02258-z OPEN Macropinocytosis requires Gal-3 in a subset of patient-derived glioblastoma stem cells Laetitia Seguin1,8, Soline Odouard2,8, Francesca Corlazzoli 2,8, Sarah Al Haddad2, Laurine Moindrot2, Marta Calvo Tardón3, Mayra Yebra4, Alexey Koval5, Eliana Marinari2, Viviane Bes3, Alexandre Guérin 6, Mathilde Allard2, Sten Ilmjärv6, Vladimir L. Katanaev 5, Paul R. Walker3, Karl-Heinz Krause6, Valérie Dutoit2, ✉ Jann N. Sarkaria 7, Pierre-Yves Dietrich2 & Érika Cosset 2 Recently, we involved the carbohydrate-binding protein Galectin-3 (Gal-3) as a druggable target for KRAS-mutant-addicted lung and pancreatic cancers. Here, using glioblastoma patient-derived stem cells (GSCs), we identify and characterize a subset of Gal-3high glio- 1234567890():,; blastoma (GBM) tumors mainly within the mesenchymal subtype that are addicted to Gal-3- mediated macropinocytosis. Using both genetic and pharmacologic inhibition of Gal-3, we showed a significant decrease of GSC macropinocytosis activity, cell survival and invasion, in vitro and in vivo. Mechanistically, we demonstrate that Gal-3 binds to RAB10, a member of the RAS superfamily of small GTPases, and β1 integrin, which are both required for macro- pinocytosis activity and cell survival. Finally, by defining a Gal-3/macropinocytosis molecular signature, we could predict sensitivity to this dependency pathway and provide proof-of- principle for innovative therapeutic strategies to exploit this Achilles’ heel for a significant and unique subset of GBM patients. 1 University Côte d’Azur, CNRS UMR7284, INSERM U1081, Institute for Research on Cancer and Aging (IRCAN), Nice, France. 2 Laboratory of Tumor Immunology, Department of Oncology, Center for Translational Research in Onco-Hematology, Swiss Cancer Center Léman (SCCL), Geneva University Hospitals, University of Geneva, Geneva, Switzerland. -

Single Cell Derived Clonal Analysis of Human Glioblastoma Links
SUPPLEMENTARY INFORMATION: Single cell derived clonal analysis of human glioblastoma links functional and genomic heterogeneity ! Mona Meyer*, Jüri Reimand*, Xiaoyang Lan, Renee Head, Xueming Zhu, Michelle Kushida, Jane Bayani, Jessica C. Pressey, Anath Lionel, Ian D. Clarke, Michael Cusimano, Jeremy Squire, Stephen Scherer, Mark Bernstein, Melanie A. Woodin, Gary D. Bader**, and Peter B. Dirks**! ! * These authors contributed equally to this work.! ** Correspondence: [email protected] or [email protected]! ! Supplementary information - Meyer, Reimand et al. Supplementary methods" 4" Patient samples and fluorescence activated cell sorting (FACS)! 4! Differentiation! 4! Immunocytochemistry and EdU Imaging! 4! Proliferation! 5! Western blotting ! 5! Temozolomide treatment! 5! NCI drug library screen! 6! Orthotopic injections! 6! Immunohistochemistry on tumor sections! 6! Promoter methylation of MGMT! 6! Fluorescence in situ Hybridization (FISH)! 7! SNP6 microarray analysis and genome segmentation! 7! Calling copy number alterations! 8! Mapping altered genome segments to genes! 8! Recurrently altered genes with clonal variability! 9! Global analyses of copy number alterations! 9! Phylogenetic analysis of copy number alterations! 10! Microarray analysis! 10! Gene expression differences of TMZ resistant and sensitive clones of GBM-482! 10! Reverse transcription-PCR analyses! 11! Tumor subtype analysis of TMZ-sensitive and resistant clones! 11! Pathway analysis of gene expression in the TMZ-sensitive clone of GBM-482! 11! Supplementary figures and tables" 13" "2 Supplementary information - Meyer, Reimand et al. Table S1: Individual clones from all patient tumors are tumorigenic. ! 14! Fig. S1: clonal tumorigenicity.! 15! Fig. S2: clonal heterogeneity of EGFR and PTEN expression.! 20! Fig. S3: clonal heterogeneity of proliferation.! 21! Fig. -

Novel Gene Signatures for Stage Classification of the Squamous Cell
www.nature.com/scientificreports OPEN Novel gene signatures for stage classifcation of the squamous cell carcinoma of the lung Angel Juarez‑Flores, Gabriel S. Zamudio & Marco V. José* The squamous cell carcinoma of the lung (SCLC) is one of the most common types of lung cancer. As GLOBOCAN reported in 2018, lung cancer was the frst cause of death and new cases by cancer worldwide. Typically, diagnosis is made in the later stages of the disease with few treatment options available. The goal of this work was to fnd some key components underlying each stage of the disease, to help in the classifcation of tumor samples, and to increase the available options for experimental assays and molecular targets that could be used in treatment development. We employed two approaches. The frst was based in the classic method of diferential gene expression analysis, network analysis, and a novel concept known as network gatekeepers. The second approach was using machine learning algorithms. From our combined approach, we identifed two sets of genes that could function as a signature to identify each stage of the cancer pathology. We also arrived at a network of 55 nodes, which according to their biological functions, they can be regarded as drivers in this cancer. Although biological experiments are necessary for their validation, we proposed that all these genes could be used for cancer development treatments. As GLOBOCAN reported in 2018, lung cancer was the frst cause of deaths and new cases by cancer worldwide 1. Squamous cell carcinoma of the lung (SCC) is one type of lung cancer which comprises approximately 30% of all lung cancer cases. -

Case-Control Study for Colorectal Cancer Genetic Susceptibility in EPICOLON: Previously Identified Variants and Mucins
UC Davis UC Davis Previously Published Works Title Case-control study for colorectal cancer genetic susceptibility in EPICOLON: previously identified variants and mucins. Permalink https://escholarship.org/uc/item/48x6z4pq Journal BMC cancer, 11(1) ISSN 1471-2407 Authors Abulí, Anna Fernández-Rozadilla, Ceres Alonso-Espinaco, Virginia et al. Publication Date 2011-08-05 DOI 10.1186/1471-2407-11-339 Peer reviewed eScholarship.org Powered by the California Digital Library University of California Abulí et al. BMC Cancer 2011, 11:339 http://www.biomedcentral.com/1471-2407/11/339 RESEARCHARTICLE Open Access Case-control study for colorectal cancer genetic susceptibility in EPICOLON: previously identified variants and mucins Anna Abulí1,2, Ceres Fernández-Rozadilla3, Virginia Alonso-Espinaco1, Jenifer Muñoz1, Victoria Gonzalo1, Xavier Bessa2, Dolors González4, Joan Clofent5, Joaquin Cubiella6, Juan D Morillas7, Joaquim Rigau8, Mercedes Latorre9, Fernando Fernández-Bañares10, Elena Peña11, Sabino Riestra12, Artemio Payá13, Rodrigo Jover13, Rosa M Xicola14, Xavier Llor14, Luis Carvajal-Carmona15, Cristina M Villanueva16, Victor Moreno17, Josep M Piqué1, Angel Carracedo3, Antoni Castells1, Montserrat Andreu2, Clara Ruiz-Ponte3 and Sergi Castellví-Bel1*, for for the Gastrointestinal Oncology Group of the Spanish Gastroenterological Association18 Abstract Background: Colorectal cancer (CRC) is the second leading cause of cancer death in developed countries. Familial aggregation in CRC is also important outside syndromic forms and, in this case, a polygenic model with several common low-penetrance alleles contributing to CRC genetic predisposition could be hypothesized. Mucins and GALNTs (N-acetylgalactosaminyltransferase) are interesting candidates for CRC genetic susceptibility and have not been previously evaluated. We present results for ten genetic variants linked to CRC risk in previous studies (previously identified category) and 18 selected variants from the mucin gene family in a case-control association study from the Spanish EPICOLON consortium. -

Table S1. 103 Ferroptosis-Related Genes Retrieved from the Genecards
Table S1. 103 ferroptosis-related genes retrieved from the GeneCards. Gene Symbol Description Category GPX4 Glutathione Peroxidase 4 Protein Coding AIFM2 Apoptosis Inducing Factor Mitochondria Associated 2 Protein Coding TP53 Tumor Protein P53 Protein Coding ACSL4 Acyl-CoA Synthetase Long Chain Family Member 4 Protein Coding SLC7A11 Solute Carrier Family 7 Member 11 Protein Coding VDAC2 Voltage Dependent Anion Channel 2 Protein Coding VDAC3 Voltage Dependent Anion Channel 3 Protein Coding ATG5 Autophagy Related 5 Protein Coding ATG7 Autophagy Related 7 Protein Coding NCOA4 Nuclear Receptor Coactivator 4 Protein Coding HMOX1 Heme Oxygenase 1 Protein Coding SLC3A2 Solute Carrier Family 3 Member 2 Protein Coding ALOX15 Arachidonate 15-Lipoxygenase Protein Coding BECN1 Beclin 1 Protein Coding PRKAA1 Protein Kinase AMP-Activated Catalytic Subunit Alpha 1 Protein Coding SAT1 Spermidine/Spermine N1-Acetyltransferase 1 Protein Coding NF2 Neurofibromin 2 Protein Coding YAP1 Yes1 Associated Transcriptional Regulator Protein Coding FTH1 Ferritin Heavy Chain 1 Protein Coding TF Transferrin Protein Coding TFRC Transferrin Receptor Protein Coding FTL Ferritin Light Chain Protein Coding CYBB Cytochrome B-245 Beta Chain Protein Coding GSS Glutathione Synthetase Protein Coding CP Ceruloplasmin Protein Coding PRNP Prion Protein Protein Coding SLC11A2 Solute Carrier Family 11 Member 2 Protein Coding SLC40A1 Solute Carrier Family 40 Member 1 Protein Coding STEAP3 STEAP3 Metalloreductase Protein Coding ACSL1 Acyl-CoA Synthetase Long Chain Family Member 1 Protein -

Unraveling the Relevance of ARL Gtpases in Cutaneous Melanoma Prognosis Through Integrated Bioinformatics Analysis
International Journal of Molecular Sciences Article Unraveling the Relevance of ARL GTPases in Cutaneous Melanoma Prognosis through Integrated Bioinformatics Analysis Cheila Brito 1, Bruno Costa-Silva 2 , Duarte C. Barral 3 and Marta Pojo 1,* 1 Unidade de Investigação em Patobiologia Molecular (UIPM) do Instituto Português de Oncologia de Lisboa Francisco Gentil E.P.E., Rua Prof. Lima Basto, 1099-023 Lisbon, Portugal; [email protected] 2 Champalimaud Research, Champalimaud Centre for the Unknown, Avenida de Brasília, 1400-038 Lisbon, Portugal; [email protected] 3 iNOVA4Health, CEDOC, NOVA Medical School, NMS, Universidade NOVA de Lisboa, 1169-056 Lisbon, Portugal; [email protected] * Correspondence: [email protected]; Tel.: +351-217-229-800 Abstract: Cutaneous melanoma (CM) is the deadliest skin cancer, whose molecular pathways underlying its malignancy remain unclear. Therefore, new information to guide evidence-based clinical decisions is required. Adenosine diphosphate (ADP)-ribosylation factor-like (ARL) proteins are membrane trafficking regulators whose biological relevance in CM is undetermined. Here, we investigated ARL expression and its impact on CM prognosis and immune microenvironment through integrated bioinformatics analysis. Our study found that all 22 ARLs are differentially expressed Citation: Brito, C.; Costa-Silva, B.; in CM. Specifically, ARL1 and ARL11 are upregulated and ARL15 is downregulated regardless Barral, D.C.; Pojo, M. Unraveling the of mutational frequency or copy number variations. According to TCGA data, ARL1 and ARL15 Relevance of ARL GTPases in represent independent prognostic factors in CM as well as ARL11 based on GEPIA and OncoLnc. Cutaneous Melanoma Prognosis To investigate the mechanisms by which ARL1 and ARL11 increase patient survival while ARL15 through Integrated Bioinformatics reduces it, we evaluated their correlation with the immune microenvironment. -

Microrna-15A and -16-1 Act Via MYB to Elevate Fetal Hemoglobin Expression in Human Trisomy 13
MicroRNA-15a and -16-1 act via MYB to elevate fetal hemoglobin expression in human trisomy 13 Vijay G. Sankarana,b,c,d, Tobias F. Mennee,f, Danilo Šcepanovicg, Jo-Anne Vergilioh, Peng Jia, Jinkuk Kima,g,i, Prathapan Thirua, Stuart H. Orkind,e,f,j, Eric S. Landera,b,k,l, and Harvey F. Lodisha,b,l,1 aWhitehead Institute for Biomedical Research, Cambridge, MA 02142; bBroad Institute of Massachusetts Institute of Technology and Harvard University, Cambridge, MA 02142; Departments of cMedicine and hPathology and eDivision of Hematology/Oncology, Children’s Hospital Boston, Boston, MA 02115; Departments of dPediatrics and kSystems Biology, Harvard Medical School, Boston, MA 02115; lDepartment of Biology and iHoward Hughes Medical Institute, Massachusetts Institute of Technology, Cambridge, MA 02142; gHarvard–Massachusetts Institute of Technology Division of Health Sciences and Technology, Cambridge, MA 02142; fDepartment of Pediatric Oncology, Dana-Farber Cancer Institute, Boston, MA 02115; and jThe Howard Hughes Medical Institute, Boston, MA 02115 Contributed by Harvey F. Lodish, December 13, 2010 (sent for review November 8, 2010) Many human aneuploidy syndromes have unique phenotypic other hematopoietic cells shifts to the bone marrow, the pre- consequences, but in most instances it is unclear whether these dominant postnatal site for hematopoiesis, another switch phenotypes are attributable to alterations in the dosage of specific occurs, resulting in down-regulation of γ-globin and concomitant genes. In human trisomy 13, there is delayed switching and up-regulation of the adult β-globin gene (6, 8, 10). There is persistence of fetal hemoglobin (HbF) and elevation of embryonic a limited understanding of the molecular control of these globin hemoglobin in newborns. -

Systems-Based Interactome Analysis for Hematopoiesis Effect of Angelicae Sinensis Radix: Regulated Network of Cell Proliferation Towards Hemopoiesis
Chin J Integr Med 2019 Dec;25(12):939-947 • 939 • Available online at link.springer.com/journal/11655 hinese Journal of Integrative Medicine Journal homepage: www.cjim.cn/zxyjhen/zxyjhen/ch/index.aspx C E-mail: [email protected] Interdisciplinary Knowledge Systems-Based Interactome Analysis for Hematopoiesis Effect of Angelicae sinensis Radix: Regulated Network of Cell Proliferation towards Hemopoiesis ZHENG Guang1, ZHANG He1, YANG Yun1, SUN Ying-li2, ZHANG Yan-jing1, CHEN Ju-ping1, HAO Ting1, LU Cheng3, GUO Hong-tao4, ZHANG Ge5, FAN Dan-ping3, HE Xiao-juan3, and LU Ai-ping5 ABSTRACT Objective: To explore the molecular-level mechanism on the hematopoiesis effect of Angelicae sinensis Radix (ASR) with systems-based interactome analysis. Methods: This systems-based interactome analysis was designed to enforce the workflow of "ASR (herb)→compound→target protein→internal protein actions→ending regulated protein for hematopoiesis". This workfl ow was deployed with restrictions on regulated proteins expresses in bone marrow and anemia disease and futher validated with experiments. Results: The hematopoiesis mechanism of ASR might be accomplished through regulating pathways of cell proliferation towards hemopoiesis with cross-talking agents of spleen tyrosine kinase (SYK), Janus kinase 2 (JAK2), and interleukin-2-inducible T-cell kinase (ITK). The hematopoietic function of ASR was also validated by colony- forming assay performed on mice bone marrow cells. As a result, SYK, JAK2 and ITK were activated. Conclusion: This study provides a new approach to systematically study and predict the therapeutic mechanism for ASR based on interactome analysis towards biological process with experimental validations. KEYWORDS Angelicae sinensis Radix, interactome, cell proliferation, hemopoiesis, regulation, cross-talking agents Angelicae sinensis Radix (ASR) is a widely the role of Janus kinase 2 (JAK2) and mothers against used Chinese herbal medicine (CHM) of the dried decapentaplegic protein (SMAD) 1/5/8 pathways roots of Angelica sinensis (Oliv.) Diels (Apiaceae). -

Computational Analysis of AP-2Γ Role in Bladder Cancer
Review and Research on Cancer Treatment Volume 6, Issue 1 (2020) Computational analysis of AP-2γ role in bladder cancer Damian Kołat1#*, Żaneta Kałuzińska1#, Elżbieta Płuciennik1 1 Department of Molecular Carcinogenesis, Medical University of Lodz, 90-752 Lodz, Poland # These authors contributed equally to the work * Corresponding author: Damian Kołat, Department of Molecular Carcinogenesis, Medical University of Lodz, e-mail: [email protected] ABSTRACT AP-2γ protein is a member of AP-2 transcription factors family, which participate in early developmental processes. Its oncogenic function has been confirmed in few cancers (e.g. breast, skin or ovarian) whereas the role in bladder cancer (BLCA) is not well understood. Nowadays, disruptions in pathways such as FGFR3/RAS or TP53/RB1 are the best known, yet new genes that would allow a better understanding of this heterogeneous tumor are being researched. The current literature indicates the promoting effect of AP-2γ encoding gene (TFAP2C) on distant recurrence or tumor growth. Similar relationship, according to which increase in TFAP2C expression significantly worsen prognostic endpoints, was observed by performing GEPIA on bladder cancer cohort. Using UALCAN platform, we noticed TFAP2C expression increase in bladder cancer compared to normal tissue. To assess the role of this gene in BLCA, GSEA analysis was performed regarding the AP-2γ targets on TFAP2C "high" and "low" groups. Beside interesting patterns related to groups distinction by genes whose expression profile is specific to BLCA, we found other curious results which we implemented for DAVID bioinformatic resources. Using functional annotation, we described selected gene sets having highest statistical significance. -
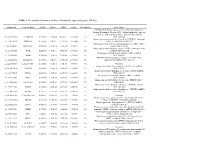
Table 1 the Statistical Metrics for Key Differentially Expressed Genes (Degs)
Table 1 The statistical metrics for key differentially expressed genes (DEGs) Agiliant Id Gene Symbol logFC pValue FDR tvalue Regulation Gene Name PREDICTED: Homo sapiens similar to Filamin-C (Gamma- filamin) (Filamin-2) (Protein FLNc) (Actin-binding-like protein) (ABP-L) (ABP-280-like protein) (LOC649056), mRNA A_24_P237896 LOC649056 0.509843 9.18E-14 4.54E-11 12.07302 Up [XR_018580] Homo sapiens programmed cell death 10 (PDCD10), transcript A_23_P18325 PDCD10 0.243111 2.8E-12 9.24E-10 10.62808 Up variant 1, mRNA [NM_007217] Homo sapiens Rho GTPase activating protein 27 (ARHGAP27), A_23_P141335 ARHGAP27 0.492709 3.97E-12 1.22E-09 10.48571 Up mRNA [NM_199282] Homo sapiens tubby homolog (mouse) (TUB), transcript variant A_23_P53110 TUB 0.528219 1.77E-11 4.56E-09 9.891033 Up 1, mRNA [NM_003320] Homo sapiens MyoD family inhibitor (MDFI), mRNA A_23_P42168 MDFI 0.314474 1.81E-10 3.74E-08 8.998697 Up [NM_005586] PREDICTED: Homo sapiens hypothetical LOC644701 A_32_P56890 LOC644701 0.444703 3.6E-10 7.09E-08 8.743973 Up (LOC644701), mRNA [XM_932316] A_32_P167111 A_32_P167111 0.873588 7.41E-10 1.4E-07 8.47781 Up Unknown Homo sapiens zinc finger protein 784 (ZNF784), mRNA A_24_P221424 ZNF784 0.686781 9.18E-10 1.68E-07 8.399687 Up [NM_203374] Homo sapiens lin-28 homolog (C. elegans) (LIN28), mRNA A_23_P74895 LIN28 0.218876 1.27E-09 2.24E-07 8.282224 Up [NM_024674] Homo sapiens ribosomal protein L5 (RPL5), mRNA A_23_P12140 RPL5 0.247598 1.81E-09 3.11E-07 8.154317 Up [NM_000969] Homo sapiens cDNA FLJ43841 fis, clone TESTI4006137. -

Supplemental Figures
Control #1 Control #2 Control #3 83972 #1 83972 #2 83972 #3 CFT073 #1 CFT073 #2 CFT073 #3 Log-intensity ratios (M-values) ratios Log-intensity Log-intensity averages (A-values) Supplemental Figure 1. MA-plots showing normalized raw data using cross- correlation. The data was found to be of high quality with high replicate correlation (correlation>0.98) and with no systematic bias. A 83972 B CFT073 Supplemental Figure 2. Inversely regulated gene networks generated by IPA. The five inversely regulated genes belonged to one functional network in which HSPA6, FOSB and RN7SK interact with TATA box binding protein (TBP), a general transcription factor required for RNA polymerase II activity. The ABU strain 83972 (A) suppressed FOSB and other genes in this network while the CFT073 strain (B) enhanced their expression. 83972 also suppressed RGS4, a negative regulator of G-protein-coupled receptor-induced signaling and IFIT1, a gene of the type I IFN signaling pathway essential for the antibacterial effector response. A No of affected Network genes Score Top functions genes BAIAP2, BANP, BRIX1, Caspase, CTNNB1, Cyclin A, DACH1, Infection Mechanisms, EMP1, EPHA4, FGFBP1, GADD45G, HES1, HIST2H2BE, IgG, Infectious Disease, IL12, Interferon α, Jnk, MAVS, MDM2, NFkB, NOD1, P38 MAPK, 51 24 Endocrine System PDCD4, PELI2, PEMT, Ras homolog, TFAP2A, TLR5, Development and TNFRSF10D, UBC, Ubiquitin, VANGL2, VARS, Vegf, ZC3HAV1 Function AHCTF1, AKR1C2, ALOX5AP, ARL4A, BAMBI, BAZ2B, Bcl9- Cbp/p300-Ctnnb1-Lef/Tcf, BRF2, CA9, Ca2+, CNN2, CRIP1, Small Molecule -

Phd Thesis by Martin Kircher
Understanding and improving high-throughput sequencing data production and analysis Der Fakult¨at fur¨ Mathematik und Informatik der Universit¨at Leipzig angenommene DISSERTATION zur Erlangung des akademischen Grades DOCTOR RERUM NATURALIUM (Dr. rer. nat.) im Fachgebiet Informatik vorgelegt von M. Sc. hon. Martin Kircher geboren am 30. September 1983 in Erfurt Die Annahme der Dissertation wurde empfohlen von: 1. Professor Dr. Anton Nekrutenko (PennState University, USA) 2. Professor Dr. Peter F. Stadler (Leipzig) Die Verleihung des akademischen Grades erfolgt mit Bestehen der Verteidigung am 11. Juli 2011 mit dem Gesamtpr¨adikat summa cum laude. Kircher, Martin Understanding and improving high-throughput sequencing data production and analysis Max-Planck Institute for Evolutionary Anthropology, Leipzig University, Germany, Dissertation 2011 226 pages, 254 references, 66 figures, 23 tables Preface Abstract Advances in DNA sequencing revolutionized the field of genomics over the last 5 years. New sequencing instruments make it possible to rapidly generate large amounts of sequence data at substantially lower cost. These high-throughput sequencing technologies (e.g. Roche 454 FLX, Life Technology SOLiD, Dover Polonator, Helicos HeliScope and Illumina Genome Analyzer) make whole genome sequencing and resequencing, transcript sequencing as well as quantification of gene expression, DNA-protein interactions and DNA methylation feasible at an unanticipated scale. In the field of evolutionary genomics, high-throughput sequencing permitted studies