REPORT Sequence Alterations Within CYP7B1 Implicate Defective Cholesterol Homeostasis in Motor-Neuron Degeneration
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Mining for Oxysterols in Cyp7b1-/- Mouse Brain and Plasma
biomolecules Communication − − Mining for Oxysterols in Cyp7b1 / Mouse Brain and Plasma: Relevance to Spastic Paraplegia Type 5 Anna Meljon 1,2, Peter J. Crick 1, Eylan Yutuc 1 , Joyce L. Yau 3, Jonathan R. Seckl 3, Spyridon Theofilopoulos 1,4 , Ernest Arenas 4, Yuqin Wang 1 and William J. Griffiths 1,* 1 Swansea University Medical School, ILS1 Building, Singleton Park, Swansea SA2 8PP, UK; [email protected] (A.M.); [email protected] (P.J.C.); [email protected] (E.Y.); s.theofi[email protected] (S.T.); [email protected] (Y.W.) 2 Institute for Global Food Security, Queens University Belfast, Stranmillis Road, Belfast BT9 5AG, UK 3 Endocrinology Unit, BHF Centre for Cardiovascular Science, The Queen’s Medical Research Institute, University of Edinburgh, 47 Little France Crescent, Edinburgh EH16 4TJ, UK; [email protected] (J.L.Y.); [email protected] (J.R.S.) 4 Laboratory of Molecular Neurobiology, Department of Medical Biochemistry and Biophysics, Karolinska Institutet, SE-17177 Stockholm, Sweden; [email protected] * Correspondence: w.j.griffi[email protected]; Tel.: +44-1792-295562 Received: 6 March 2019; Accepted: 2 April 2019; Published: 13 April 2019 Abstract: Deficiency in cytochrome P450 (CYP) 7B1, also known as oxysterol 7α-hydroxylase, in humans leads to hereditary spastic paraplegia type 5 (SPG5) and in some cases in infants to liver disease. SPG5 is medically characterized by loss of motor neurons in the corticospinal tract. In an effort to gain a better understanding of the fundamental biochemistry of this disorder, we have extended our previous profiling of the oxysterol content of brain and plasma of Cyp7b1 knockout (-/-) mice to include, amongst other sterols, 25-hydroxylated cholesterol metabolites. -

Polyclonal Antibody to CYP7B1 (C-Term) - Aff - Purified
OriGene Technologies, Inc. OriGene Technologies GmbH 9620 Medical Center Drive, Ste 200 Schillerstr. 5 Rockville, MD 20850 32052 Herford UNITED STATES GERMANY Phone: +1-888-267-4436 Phone: +49-5221-34606-0 Fax: +1-301-340-8606 Fax: +49-5221-34606-11 [email protected] [email protected] AP20144PU-N Polyclonal Antibody to CYP7B1 (C-term) - Aff - Purified Alternate names: 25-hydroxycholesterol 7-alpha-hydroxylase, Cytochrome P450 7B1, Oxysterol 7-alpha- hydroxylase Quantity: 0.1 mg Concentration: 0.5 mg/ml Background: P450 enzymes constitute a family of monooxygenase enzymes that are involved in the metabolism of a wide array of endogenous and xenobiotic compounds including cholesterol. CYP8B1 moderates the ratio of cholic acid over chenodeoxycholic acid to control the solubility of cholesterol. P450 cholesterol 7-hydroxylase, CYP7A1, is the rate limiting enzyme of bile acid synthesis in the liver, and its expression is mediated by the bile acid receptor FXR. CYP27A1 catalyzes vitamin D3 25-hydroxylation and is localized to the mitochondria in kidney and liver. CYP7B1 (oxysterol 7-α-hydroxylase) functions as an enzyme in the alternate bile acid synthesis pathway. Specifically, CYP7B1 metabolizes 25-and 27-hydroxycholesterol. The gene encoding human CYP7B1 maps to chromosome 8q21.3. Mutations in the CYP7B1 gene may cause a metabolic defect in bile acid synthesis characterized by elevated urinary bile acid excretion, severe cholestasis, cirrhosis and liver synthetic failure. Uniprot ID: O75881 NCBI: NP_004811 GeneID: 9420 Host: Goat Immunogen: Peptide with sequence C-YPDSDVLFRYKVKS, from the C Terminus of the protein sequence according to NP_004811.1. Format: State: Liquid purified IgG fraction. -

Alteration of the Steroidogenesis in Boys with Autism Spectrum Disorders
Janšáková et al. Translational Psychiatry (2020) 10:340 https://doi.org/10.1038/s41398-020-01017-8 Translational Psychiatry ARTICLE Open Access Alteration of the steroidogenesis in boys with autism spectrum disorders Katarína Janšáková 1, Martin Hill 2,DianaČelárová1,HanaCelušáková1,GabrielaRepiská1,MarieBičíková2, Ludmila Máčová2 and Daniela Ostatníková1 Abstract The etiology of autism spectrum disorders (ASD) remains unknown, but associations between prenatal hormonal changes and ASD risk were found. The consequences of these changes on the steroidogenesis during a postnatal development are not yet well known. The aim of this study was to analyze the steroid metabolic pathway in prepubertal ASD and neurotypical boys. Plasma samples were collected from 62 prepubertal ASD boys and 24 age and sex-matched controls (CTRL). Eighty-two biomarkers of steroidogenesis were detected using gas-chromatography tandem-mass spectrometry. We observed changes across the whole alternative backdoor pathway of androgens synthesis toward lower level in ASD group. Our data indicate suppressed production of pregnenolone sulfate at augmented activities of CYP17A1 and SULT2A1 and reduced HSD3B2 activity in ASD group which is partly consistent with the results reported in older children, in whom the adrenal zona reticularis significantly influences the steroid levels. Furthermore, we detected the suppressed activity of CYP7B1 enzyme readily metabolizing the precursors of sex hormones on one hand but increased anti-glucocorticoid effect of 7α-hydroxy-DHEA via competition with cortisone for HSD11B1 on the other. The multivariate model found significant correlations between behavioral indices and circulating steroids. From dependent variables, the best correlation was found for the social interaction (28.5%). Observed changes give a space for their utilization as biomarkers while reveal the etiopathogenesis of ASD. -

Expression of CYP2S1 in Human Hepatic Stellate Cells
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Elsevier - Publisher Connector FEBS Letters 581 (2007) 781–786 Expression of CYP2S1 in human hepatic stellate cells Carylyn J. Mareka, Steven J. Tuckera, Matthew Korutha, Karen Wallacea,b, Matthew C. Wrighta,b,* a School of Medical Sciences, Institute of Medical Science, University of Aberdeen, Aberdeen, UK b Liver Faculty Research Group, School of Clinical and Laboratory Sciences, University of Newcastle, Newcastle, UK Received 22 November 2006; revised 16 January 2007; accepted 23 January 2007 Available online 2 February 2007 Edited by Laszlo Nagy the expression and accumulation of scarring extracellular Abstract Activated stellate cells are myofibroblast-like cells associated with the generation of fibrotic scaring in chronically fibrotic matrix protein [2]. It is currently thought an inhibition damaged liver. Gene chip analysis was performed on cultured fi- of fibrosis in liver diseases may be an effective approach to brotic stellate cells. Of the 51 human CYP genes known, 13 treating patients for which the cause is refractive to current CYP and 5 CYP reduction-related genes were detected with 4 treatments (e.g. in approx. 30% of hepatitis C infected CYPs (CYP1A1, CYP2E1, CY2S1 and CYP4F3) consistently patients) [2,3]. At present, there is no approved treatment for present in stellate cells isolated from three individuals. Quantita- fibrosis. tive RT-PCR indicated that CYP2S1 was a major expressed Inadvertent toxicity of drugs is often associated with a ‘‘met- CYP mRNA transcript. The presence of a CYP2A-related pro- abolic activation’’ by CYPs [1]. -

Glyphosate's Suppression of Cytochrome P450 Enzymes
Entropy 2013, 15, 1416-1463; doi:10.3390/e15041416 OPEN ACCESS entropy ISSN 1099-4300 www.mdpi.com/journal/entropy Review Glyphosate’s Suppression of Cytochrome P450 Enzymes and Amino Acid Biosynthesis by the Gut Microbiome: Pathways to Modern Diseases Anthony Samsel 1 and Stephanie Seneff 2,* 1 Independent Scientist and Consultant, Deerfield, NH 03037, USA; E-Mail: [email protected] 2 Computer Science and Artificial Intelligence Laboratory, MIT, Cambridge, MA 02139, USA * Author to whom correspondence should be addressed; E-Mail: [email protected]; Tel.: +1-617-253-0451; Fax: +1-617-258-8642. Received: 15 January 2013; in revised form: 10 April 2013 / Accepted: 10 April 2013 / Published: 18 April 2013 Abstract: Glyphosate, the active ingredient in Roundup®, is the most popular herbicide used worldwide. The industry asserts it is minimally toxic to humans, but here we argue otherwise. Residues are found in the main foods of the Western diet, comprised primarily of sugar, corn, soy and wheat. Glyphosate's inhibition of cytochrome P450 (CYP) enzymes is an overlooked component of its toxicity to mammals. CYP enzymes play crucial roles in biology, one of which is to detoxify xenobiotics. Thus, glyphosate enhances the damaging effects of other food borne chemical residues and environmental toxins. Negative impact on the body is insidious and manifests slowly over time as inflammation damages cellular systems throughout the body. Here, we show how interference with CYP enzymes acts synergistically with disruption of the biosynthesis of aromatic amino acids by gut bacteria, as well as impairment in serum sulfate transport. Consequences are most of the diseases and conditions associated with a Western diet, which include gastrointestinal disorders, obesity, diabetes, heart disease, depression, autism, infertility, cancer and Alzheimer’s disease. -
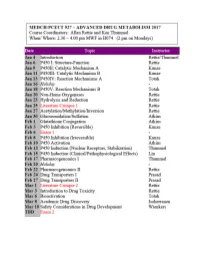
CYTOCHROME P450: Structure-Function
MEDCH 527 AER Jan. 4-6, 2017 CYTOCHROME P450: Structure-Function 1. General P450 Characteristics anD Taxonomy 2. Human P450s – Substrate anD Inhibitor Selectivities 3. Structure-Function Aspects of LiganD BinDing, P450 ReDuction anD Oxygen Activation References P450 Homepage -http:// http://drnelson.uthsc.edu/CytochromeP450.html Nelson DR et al., Pharmacogenetics. 1996 6(1):1-42: Nelson DR et al., Pharmacogenetics. 2004 14(1):1-18. Testa, B. The Biochemistry of Drug Metabolism: A 6 Part Series in Chem. BioDivers. (2006-2008). Sligar, SG. Glimpsing the critical intermediate in cytochrome P450 oxidations. Science. 2010 Nov 12;330(6006):924-5. Johnson EF, et al. Correlating structure and function of drug metabolizing enzymes:Progress and ongoing challenges. Drug Metab. Dispos. 42:9-22 (2014). Zientek MA and Youdim K, Reaction Phenotyping:Advances in experimental strategies used to characterize the contribution of drug metabolizing enzymes. Drug Metab. Dispos. 43:163-181 (2015). Almazroo et al., Drug Metabolism in the Liver. Clin. Liver Dis. 21:1-20 (2017). Absorption, Distribution, Metabolism and Excretion (ADME) of Orally Administered Drugs To reach their sites of action in the body, orally administered drugs must be absorbed from the small intestine, survive first pass metabolism - typically in the liver - before eventually being excreted, usually as drug metabolites in the bile and kidney. Therefore, clinically useful drugs must be able to cross an array of cell membranes, which are composed of a lipid bilayer. Drugs must exhibit an adequate degree of lipophilicity (logP of ~2-4) in order to able to dissolve into this lipoidal environment. Many drug metabolism processes render lipophilic drugs more water-soluble so as to facilitate excretion via the kidneys and bile. -

Biodiversity of P-450 Monooxygenase: Cross-Talk
Cytochrome P450: Oxygen activation and biodiversty 1 Biodiversity of P-450 monooxygenase: Cross-talk between chemistry and biology Heme Fe(II)-CO complex 450 nm, different from those of hemoglobin and other heme proteins 410-420 nm. Cytochrome Pigment of 450 nm Cytochrome P450 CYP3A4…. 2 High Energy: Ultraviolet (UV) Low Energy: Infrared (IR) Soret band 420 nm or g-band Mb Fe(II) ---------- Mb Fe(II) + CO - - - - - - - Visible region Visible bands Q bands a-band, b-band b a 3 H2O/OH- O2 CO Fe(III) Fe(II) Fe(II) Fe(II) Soret band at 420 nm His His His His metHb deoxy Hb Oxy Hb Carbon monoxy Hb metMb deoxy Mb Oxy Mb Carbon monoxy Mb H2O/Substrate O2-Substrate CO Substrate Soret band at 450 nm Fe(III) Fe(II) Fe(II) Fe(II) Cytochrome P450 Cys Cys Cys Cys Active form 4 Monooxygenase Reactions by Cytochromes P450 (CYP) + + RH + O2 + NADPH + H → ROH + H2O + NADP RH: Hydrophobic (lipophilic) compounds, organic compounds, insoluble in water ROH: Less hydrophobic and slightly soluble in water. Drug metabolism in liver ROH + GST → R-GS GST: glutathione S-transferase ROH + UGT → R-UG UGT: glucuronosyltransferaseGlucuronic acid Insoluble compounds are converted into highly hydrophilic (water soluble) compounds. 5 Drug metabolism at liver: Sleeping pill, pain killer (Narcotic), carcinogen etc. Synthesis of steroid hormones (steroidgenesis) at adrenal cortex, brain, kidney, intestine, lung, Animal (Mammalian, Fish, Bird, Insect), Plants, Fungi, Bacteria 6 NSAID: non-steroid anti-inflammatory drug 7 8 9 10 11 Cytochrome P450: Cysteine-S binding to Fe(II) heme is important for activation of O2. -

Spatiotemporal Dynamics of the Expression of Estrogen Receptors In
Molecular Psychiatry (2009) 14, 223–232 & 2009 Nature Publishing Group All rights reserved 1359-4184/09 $32.00 www.nature.com/mp ORIGINAL ARTICLE Spatiotemporal dynamics of the expression of estrogen receptors in the postnatal mouse brain N Sugiyama1, S Andersson1, R Lathe2, X Fan1, P Alonso-Magdalena1, T Schwend1, I Nalvarte1, M Warner1 and J-A˚ Gustafsson1 1Department of Biosciences and Nutrition, Karolinska Institute, Novum, Huddinge, Sweden and 2Pieta Research, Edinburgh, UK This study reports on the spatiotemporal dynamics of the expression of estrogen receptors (ERs) in the mouse central nervous system (CNS) during the early postnatal and the peripubertal period. At postnatal day 7 (P7), neurons with strong nuclear immunostaining for both ERa and ERb1 were widely distributed throughout the brain. Sucrose density gradient sedimentation followed by western blotting supported the histochemical evidence for high levels of both ERs at P7. Over the following 2 days, there was a rapid downregulation of ERs. At P9, ERa expression was visible only in the hypothalamic area. Decline in ERb1 expression was slower than that of ERa, and ERa-negative, ERb1-positive cells were observed in the dentate gyrus and walls of third ventricle. Between P14 and P35, ERs were undetectable except for the hypothalamic area. As before P7, the ovary does not produce estrogen but does produce 5a-androstane-3b,17b-diol (3bAdiol), an estrogenic metabolite of dihydrotestosterone, we examined the effects of high levels of 3bAdiol in the postnatal period. We used CYP7B1 knockout mice which cannot hydroxylate and inactivate 3bAdiol. The brains of these mice are abnormally large with reduced apoptosis. -

Mammalian Cells Engineered to Produce Novel Steroids Emma S
bioRxiv preprint doi: https://doi.org/10.1101/261362; this version posted February 7, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Mammalian Cells Engineered to Produce Novel Steroids Emma S. Spady†,‡,§, Thomas P. Wyche‖, Nathanael J. Rollins†,§, Jon Clardy‖, Jeffrey C. Way†,§, and Pamela A. Silver*,†,§ †Department of Systems Biology, Harvard Medical School – Boston, MA 02115, United States. ‡Laboratory of Systems Pharmacology, Harvard University – Boston, MA 02115, United States. §Wyss Institute for Biologically Inspired Engineering, Harvard University – Boston, MA 02115, United States. ‖Department of Biological Chemistry and Molecular Pharmacology, Harvard Medical School – Boston, MA 02115, United States. Author Information Corresponding Author *E-mail: [email protected] Current Affiliations Thomas P. Wyche: Merck & Co., Inc., Cambridge, MA 02141, United States. 1 bioRxiv preprint doi: https://doi.org/10.1101/261362; this version posted February 7, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Abstract Steroids can be difficult to modify via traditional organic synthesis methods, but many enzymes regio- and stereo-selectively process a wide variety of steroid substrates. We tested whether steroid-modifying enzymes could make novel steroids from non-native substrates. Numerous genes encoding steroid-modifying enzymes, including some bacterial enzymes, were expressed in mammalian cells by transient transfection and found to be active. -

Hepatic Cytochrome P450 Reductase-Null Mice As an Animal Model to Study Electron Transfer Pathways in Cholesterol Synthesis and Cyp2e1-Mediated Drug Metabolism
University of Kentucky UKnowledge University of Kentucky Doctoral Dissertations Graduate School 2006 HEPATIC CYTOCHROME P450 REDUCTASE-NULL MICE AS AN ANIMAL MODEL TO STUDY ELECTRON TRANSFER PATHWAYS IN CHOLESTEROL SYNTHESIS AND CYP2E1-MEDIATED DRUG METABOLISM Li Li University of Kentucky, [email protected] Right click to open a feedback form in a new tab to let us know how this document benefits ou.y Recommended Citation Li, Li, "HEPATIC CYTOCHROME P450 REDUCTASE-NULL MICE AS AN ANIMAL MODEL TO STUDY ELECTRON TRANSFER PATHWAYS IN CHOLESTEROL SYNTHESIS AND CYP2E1-MEDIATED DRUG METABOLISM" (2006). University of Kentucky Doctoral Dissertations. 417. https://uknowledge.uky.edu/gradschool_diss/417 This Dissertation is brought to you for free and open access by the Graduate School at UKnowledge. It has been accepted for inclusion in University of Kentucky Doctoral Dissertations by an authorized administrator of UKnowledge. For more information, please contact [email protected]. ABSTRACT OF DISSERTATION Li Li The Graduate School University of Kentucky 2006 HEPATIC CYTOCHROME P450 REDUCTASE-NULL MICE AS AN ANIMAL MODEL TO STUDY ELECTRON TRANSFER PATHWAYS IN CHOLESTEROL SYNTHESIS AND CYP2E1-MEDIATED DRUG METABOLISM ABSTRACT OF DISSERTATION A dissertation submitted in partial fulfillment of the requirements of the degree of Doctor of Philosophy in The Graduate School at the University of Kentucky By Li Li Lexington, Kentucky Director: Dr. Todd D. Porter, Associate Professor of Pharmaceutical Sciences College of Pharmacy 2006 Copyright © Li Li 2006 ABSTRACT OF DISSERTATION HEPATIC CYTOCHROME P450 REDUCTASE-NULL MICE AS AN ANIMAL MODEL TO STUDY ELECTRON TRANSFER PATHWAYS IN CHOLESTEROL SYNTHESIS AND CYP2E1-MEDIATED DRUG METABOLISM NADPH-cytochrome P450 reductase (CPR) is a flavoprotein containing both FAD and FMN and functions as the electron donor protein for several oxygenase enzymes found on the endoplasmic reticulum of eukaryotic cells, including cytochrome P450s involved in drug metabolism and cholesterol biosynthesis. -

Orphan Nuclear Receptor Rorα Regulates Enzymatic Metabolism of Cerebral 24S-Hydroxycholesterol Through CYP39A1 Intronic Response Element Activation
International Journal of Molecular Sciences Article Orphan Nuclear Receptor RORα Regulates Enzymatic Metabolism of Cerebral 24S-Hydroxycholesterol through CYP39A1 Intronic Response Element Activation Hiroshi Matsuoka 1,* , Miyu Katayama 1, Ami Ohishi 1, Kaoruko Miya 1, Riki Tokunaga 1, Sou Kobayashi 1, Yuya Nishimoto 1, Kazutake Hirooka 2 , Akiho Shima 1 and Akihiro Michihara 1 1 Laboratory of Genome Function and Pathophysiology, Faculty of Pharmacy and Pharmaceutical Sciences, Fukuyama University, Fukuyama, Hiroshima 729-0292, Japan; [email protected] (M.K.); [email protected] (A.O.); [email protected] (K.M.); [email protected] (R.T.); [email protected] (S.K.); [email protected] (Y.N.); [email protected] (A.S.); [email protected] (A.M.) 2 Department of Biotechnology, Faculty of Life Science and Biotechnology, Fukuyama University, Fukuyama, Hiroshima 729-0292, Japan; [email protected] * Correspondence: [email protected]; Tel.: +81-84-936-2111 Received: 2 April 2020; Accepted: 6 May 2020; Published: 7 May 2020 Abstract: Oxysterols, important regulators of cholesterol homeostasis in the brain, are affected by neurodegenerative diseases. Early-onset Alzheimer’s disease is associated with higher levels of circulating brain-derived 24S-hydroxycholesterol (24S-OHC). Conversion of cholesterol to 24S-OHC is mediated by cholesterol 24S-hydroxylase in the brain, which is the major pathway for oxysterol elimination, followed by oxidation through hepatic first-pass metabolism by CYP39A1. Abnormal CYP39A1 expression results in accumulation of 24S-OHC, influencing neurodegenerative disease-related deterioration; thus, it is important to understand the normal elimination of 24S-OHC and the system regulating CYP39A1, a selective hepatic metabolic enzyme of 24S-OHC. -

Differential Expression of Prostaglandin I2 Synthase Associated with Arachidonic Acid Pathway in the Oral Squamous Cell Carcinoma
Hindawi Journal of Oncology Volume 2018, Article ID 6301980, 13 pages https://doi.org/10.1155/2018/6301980 Research Article Differential Expression of Prostaglandin I2 Synthase Associated with Arachidonic Acid Pathway in the Oral Squamous Cell Carcinoma Anelise Russo ,1 Patr-cia M. Biselli-Chicote ,1 Rosa S. Kawasaki-Oyama,1 Márcia M. U. Castanhole-Nunes,1 José V. Maniglia,2 Dal-sio de Santi Neto,3 Érika C. Pavarino ,1 and Eny M. Goloni-Bertollo 1 1 Department of Molecular Biology: Biological and Genetics and Molecular Biology Research Unit – UPGEM, Sao˜ Jose´ do Rio Preto Medical School – FAMERP, Sao˜ Jose´ do Rio Preto, SP 15090-000, Brazil 2Department of Otorhinolaryngology and Head and Neck Surgery, FAMERP, Sao˜ Jose´ do Rio Preto, SP 15090-000, Brazil 3Department of Pathology, FAMERP, Sao˜ Jose´ do Rio Preto, SP 15090-000, Brazil Correspondence should be addressed to Anelise Russo; [email protected] and Eny M. Goloni-Bertollo; [email protected] Received 17 July 2018; Accepted 16 October 2018; Published 8 November 2018 Academic Editor: Tomas R. Chauncey Copyright © 2018 Anelise Russo et al. Tis is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Introduction. Diferential expression of genes encoding cytochrome P450 (CYP) and other oxygenases enzymes involved in biotransformation mechanisms of endogenous and exogenous compounds can lead to oral tumor development. Objective.We aimed to identify the expression profle of these genes, searching for susceptibility biomarkers in oral squamous cell carcinoma.