Phenotyping and Molecular Characterization of Proteus Vulgaris Isolated from Patients with Urinary Tract Infections
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Study of Some Virulence Factors of Proteus Mirabilis Isolated from Urinary Stones Patients
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by International Institute for Science, Technology and Education (IISTE): E-Journals Journal of Biology, Agriculture and Healthcare www.iiste.org ISSN 2224-3208 (Paper) ISSN 2225-093X (Online) Vol.5, No.23, 2015 Study of Some Virulence Factors of Proteus mirabilis Isolated from Urinary Stones Patients Mohanad J. Al-Dawah 1* Adnan H. Al-Hamadany 2 Eman M. Al-Jarallah 3 1. Al-Qasim Green University, College of Biotechnology-Genetic Engineering Department, Al-Qasim city, Babylon, Iraq 2. Al-Qadissiya University, College of Medicine-Microbiology Department 3. Babylon University, College of Science-Biology Department Abstract A total of 125 specimens of stones and urine were collected from urinary stone patients from (June to December, 2012). According to primary identification, which based on macroscopic and microscopic characteristics and biochemical tests, 25 (20%) and 100 (80%) of isolates were identified as Proteus and non-Proteus , respectively. The 25 Proteus isolates were finally identified as Proteus mirabilis based on Vitek 2 system and polymerase chain reaction (PCR) technique by using target gene 16S rRNA . The multiple logistic regressions results showed that the age ˃ 40 years old was a risk factors that significantly associated with increased incidence of P. mirabilis in urinary stone patients, as P = 0.02, and Odd’s ratio (OR) was 4.889 (1.7-14.057), while in relation to gender, the analysis revealed that they were statistically non-significant as OR was 1.174 (0.488-2.822) as well as P=0.720. -

Isolation, Identification & Characterization
Indian J Med Res 135, March 2012, pp 341-345 Isolation, identification & characterization ofProteus penneri - a missed rare pathogen Janak Kishore Department of Microbiology, Sanjay Gandhi Post-Graduate Institute of Medical Sciences, Lucknow, India Received March 4, 2010 Background & objectives: Indole negative Proteus species are invariably incorrectly identified as P. mirabilis, missing isolates of Proteus penneri. P. penneri is an invasive pathogen capable of causing major infectious diseases still seldom reported in individual cases. We report here the isolation, differentiation, characterization and typing of P. penneri from patients with different clinical infections. Methods: Urine, pus and body fluids collected from patients in intensive care units, wards and out patients departments of a tertiary health care institute from north India were cultured. A total of 61 indole negative Proteus isolates were subjected to extended biochemical tests to differentiate and identify P. penneri from P. mirabilis including failure to produce ornithine decarboxylase (by 0% strains of P. penneri and 100% strains of P. mirabilis) besides P. penneri being uniformly salicin negative, non-utilizer of citrate but ferments sucrose and maltose. Antibiograms and Dienes phenomenon were performed to characterize and type P. penneri isolates besides screening for β-lactamase production. Results: Eight isolates of P. penneri were identified; four from urine, three from abdominal drain-fluid and one from diabetic foot ulcer. P. penneri was isolated as the sole pathogen in all patients having underlying disease; post-operatively. Swarming was not seen in the first strain on primary isolation and was poor in strain-4. All eight isolates were biochemically homologous but multi-drug resistant (MDR) with resistance to 6-8 drugs (up to 12). -

Anti-Proteus Vulgaris LPS Antibody (SAB4200850)
Anti-Proteus vulgaris LPS antibody Mouse monoclonal, Clone P.vul-111 purified from hybridoma cell culture Product Number SAB4200850 Product Description In the Proteus spp. group, P. mirabilis is encountered in Monoclonal Anti-Proteus vulgaris LPS antibody (mouse the community and causes the majority of urinary tract IgM isotype) is derived from the P.vul-111 hybridoma, Proteus spp. infections, whereas P. vulgaris and produced by the fusion of mouse myeloma cells and P. penneri are less common and mainly associated with splenocytes from a mouse immunized with nosocomial none urinary infections.2,4 UV-inactivated P. vulgaris OX19 bacteria (ATCC 6380) as immunogen. The isotype is determined by ELISA P. vulgaris has a number of putative virulence factors, using Mouse Monoclonal Antibody Isotyping Reagents including the secreted hemolytic haemolysin and (Product Number ISO2). The antibody is purified from urease, which has been suggested to contribute to host culture supernatant of hybridoma cells. cell invasion, cytotoxicity, and bacterial ability to invade uroepithelial cells.4 P. vulgaris, P. mirabilis and Monoclonal Anti-Proteus vulgaris LPS specifically P. penneri harbor resistance to -lactam antibiotics as it recognizes P. vulgaris whole extract and P. vulgaris is capable of producing inducible -lactamases that LPS, the antibody has no cross reactivity with whole hydrolyze primary and extended-spectrum penicillins extract of Proteus mirabilis, P. gingivalis, E. coli, and cephalosporins. 5-6 Pseudomonas aeruginosa, Shigella flexneri, Staphylococcus aureus, or Salmonella enterica. The Reagent antibody is recommended to be used in various Supplied as a solution in 0.01 M phosphate buffered immunological techniques, including immunoblot and saline, pH 7.4, containing 15 mM sodium azide as a ELISA. -

Five Clinical Strains of P. Pettneri Were Obtained from Dr. H. C. NEU of Columbia University, NY, and from American Type Culture Collection
938 THE JOURNAL OF ANTIBIOTICS JULY 1986 PURIFICATION AND PROPERTIES OF A B-LACTAMASE FROM PROTEUS PENNERI M. E. GRACE, F. J. GREGORY, P. P. HUNG and K. P. Fu* Microbiology Division, Wyeth Labs., Inc. P.O. Box 8299, Philadelphia, PA 19101, U.S.A. (Received for publication December 9, 1985) A cephalosporin-hydrolyzing enzyme from strains of Proteus penneri resistant to B-lactam antibiotics was purified and characterized. The enzyme gave a single protein band on SDS- polyacrylamide gel electrophoresis with a molecular weight of 30,000. This cephalosporinase has an isoelectric point of 6.8, a pH optimum of 6.5 and a temperature optimum of 45°C. The enzyme hydrolyzed cephaloridine, cephalothin, cefuroxime, and cefotaxime more rapidly than penicillins. The relative rate, with cephaloridine as 100, were: cephalothin, 50; cefuroxime, 93; cefotaxime, 48; ceftriaxone, 23; cefoperazone, 11; benzylpenicillin, 3; am- picillin, 9; and carbenicillin, <1. Cephamycins had low affinities for the enzyme. However, clavulanic acid and sulbactam, with high affinites for the enzyme, were inhibitors of this enzyme. Proteus penneri has been recognized recently as a new member of the species Proteeae1). It is indole, esculin, and salicin negative, and chloramphenicol-resistant, and has been called Proteus vulgaris indole-negative or P. vulgaris biogroup 1. In a recent report, P. penneri strains were found to be more resistant to newer semi-synthetic penicillins and quinolones than P. vulgaris2,3,4). Production of 3- lactamase has been considered to be one of the important biochemical mechanisms of resistance to ,3-lactam antibiotics in bacteria5,6). YOTSUJI et al.7) and MATSUBARA et al.8) reported on a i3-lactamase from P. -

Phenotypic Detection and Antibiogram of Β‑Lactamase‑Producing Proteus Species in a Tertiary Care Hospital, India
[Downloaded free from http://www.amhsr.org] Original Article Phenotypic Detection and Antibiogram of β‑lactamase‑producing Proteus Species in a Tertiary Care Hospital, India Pal N, Hooja S, Sharma R, Maheshwari RK Department of Microbiology, SMS Medical College, Jaipur, Rajasthan, India Address for correspondence: Abstract Dr. Pal N, 82, Green Nagar, Durgapura, Jaipur, Background: Proteus species cause a variety of community‑ and hospital‑acquired illnesses. Rajasthan, India. Synthesis of β‑lactamases is the predominant mechanism for resistance to β‑lactam antibiotics. E‑mail: [email protected] Among the β‑lactamases, extended spectrum β‑lactamases (ESBLs) and AmpC β‑lactamases are the most common. Aim: The objective of this study was to determine the occurrence of ESBL and AmpC β‑lactamases in Proteus species among various clinical isolates at a tertiary care hospital, India. Materials and Methods: This study was done to identify various species of Proteus from clinical samples (n = 3922). Antimicrobial susceptibility was performed by Kirby–Bauer disc diffusion method. ESBL production was detected by modified double‑disc synergy test and indirect modified three‑dimensional tests and AmpC β‑lactamase production by AmpC disc test and modified Hodge test. Results: Proteus species were isolated in 5.4% (101/1876) specimens. Three Proteus species isolated were Proteus mirabilis 62.4% (63/101), Proteus vulgaris 29.7% (30/101), and Proteus penneri 7.9% (8/101). ESBL producers confirmed by both tests were of 88.1% (89/101). Only AmpC β‑lactamase was produced by four isolates. Coproduction of ESBL and AmpC β‑lactamase was observed in 58.4% (52/89) of isolates. -

The Production of Hlya Toxin by Proteus Penneri Strains
J. Med. Microbiol. - Vol. 39 (1993), 282-289 0 1993 The Pathological Society of Great Britain and Ireland The production of HlyA toxin by Proteus penneri strains B. W. SENIOR Department of Medical Microbiology, University of Dundee Medical School, Ninewells Hospital, Dundee DD I 9SY Summary. Twelve diverse strains of Proteus penneri of clinical origin all produced a calcium- dependent haemolysin, unlike most other Proteus spp. In most strains the haemolysin was secreted into the medium during early exponential growth and lysed not only of a variety of erythrocyte types from several animals including man, but also human neutrophils and human embryo lung fibroblasts. The haemolysin was a protein of 107 kDa, the same size as Escherichia coli HlyA, and it reacted with antiserum to E. coli HlyA. Because of its similarity in size, antigenicity and range of action to the HlyA virulence factor of E. coli, P. penneri HlyA is believed to be an important virulence factor for this organism. It was degradable by an EDTA-sensitive protease-probably the IgA protease-to inactive fragments. The interaction of P. penneri HlyA and IgA protease in vivo and the origin of HlyA, which has now been found in many diverse bacteria, are discussed. Introduction urinary tractg and wounds’O and has been isolated from the faeces of both healthy and sick individuals.” RTX toxins are a family of calcium-dependent, Potential virulence factors already known to be pro- pore-forming, cytolytic toxins produced by diverse duced by P. penneri include urease,” IgA protease13*l4 bacteria that act on different cells in different hosts. -

Proteus Infections
P.O. Box 131375, Bryanston, 2074 Ground Floor, Block 5 Bryanston Gate, Main Road Bryanston, Johannesburg, South Africa www.thistle.co.za Tel: +27 (011) 463 3260 Fax: +27 (011) 463 3036 e-mail : [email protected] Please read this bit first The HPCSA and the Med Tech Society have confirmed that this clinical case study, plus your routine review of your EQA reports from Thistle QA, should be documented as a “Journal Club” activity. This means that you must record those attending for CEU purposes. Thistle will not issue a certificate to cover these activities, nor send out “correct” answers to the CEU questions at the end of this case study. The Thistle QA CEU No is: MT00025. Each attendee should claim THREE CEU points for completing this Quality Control Journal Club exercise, and retain a copy of the relevant Thistle QA Participation Certificate as proof of registration on a Thistle QA EQA. Cycle 24 - Organism 3: Proteus Infections Proteus species are part of the Enterobacteriaceae family of gram-negative bacilli. Proteus species are most commonly found in the human intestinal tract as part of normal human intestinal flora, along with Escherichia coli and Klebsiella species. Proteus is also found in multiple environmental habitats, including long-term care facilities and hospitals. In hospital settings, it is not unusual for gram-negative bacilli to colonize both the skin and oral mucosa of both patients and hospital personnel. Infection primarily occurs from these reservoirs. Proteus mirabilis causes 90% of Proteus infections and can be considered a community-acquired infection. Proteus vulgaris and Proteus penneri are easily isolated from individuals in long-term care facilities and hospitals and from patients with underlying diseases or compromised immune systems. -
A Format for Policies and Procedures
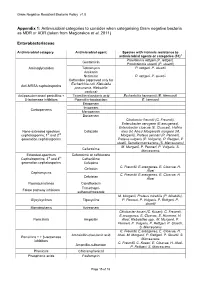
Gram Negative Resistant Bacteria Policy v1.0 Appendix 1: Antimicrobial categories to consider when categorising Gram negative bacteria as MDR or XDR (taken from Magiorakos et al, 2011) Enterobacteriaceae Antimicrobial category Antimicrobial agent Species with intrinsic resistance to antimicrobial agents or categories (51)a Providencia rettgeri (P. rettgeri), Gentamicin Provindencia stuartii (P. stuartii) Aminoglycosides Tobramycin P. rettgeri, P. stuartii Amikacin Netilmicin P. rettgeri, P. stuartii Ceftaroline (approved only for Escherichia coli, Klebsiella Anti-MRSA cephalosporins pneumonia, Klebsiella oxytoca) Antipseudomonaol penicillins + Ticarcillin-clavulanic acid Escherichia hermannii (E. Hermanii) β-lactamase inhibitors Pipercillin-taxobactam E. hermanii Ertapenem Imipenem Carbapenems Meropenem Doripenem Citrobactor freundii (C. Freundii), Enterobacter aerogene (E.areogenes), Enterobacter cloacae (E. Cloacae), Hafnia None-extended spectrum Cefazolin alvei (H. Alvei) Morganella morganii (M. cephalosporins; 1st and 2nd Morganii), Proteus penneri (P. Penneri), generation cephalosporins Proteus vulgaris (P. Vulgaris), P. Rettgeri, P stuatii, Serratia marcescens (S. Marcescens) M. Morganii, P. Penneri, P. Vulgaris, S. Cefuroxime Marcescens Extended-spectrum Cefuroxime or ceftriaxone Cephalosporins; 3rd and 4th Ceftazidime generation cephalosporins Cefepime C. Freundii, E.areogenes, E. Cloacae, H. Cefoxitin Alvei Cephamycins C. Freundii, E.areogenes, E. Cloacae, H. Cefotetan Alvei Fluoroquinolones Ciprofloxacin Trimethopri- Folate pathway inhibitors sulhamethoxazole M. Morganii, Proteus mirabilis (P. Mirabilis), Glycylcyclines Tigecycline P. Penneri, P. Vulgaris, P. Rettgeri, P. stuartii Monobactams Aztreonam Citrobacter koseri (C. Koseri), C. Freundii, E.areogenes, E. Cloacae, E. Hermanii, H. Penicillin’s Ampicillin Alvei, Klebsiellae spp., M. Morganii, P. Penneri, P. Vulgaris, P. Rettgeri, P. Stuartii, S. Marcescens C. Freundii, E.areogenes, E. Cloacae, H. Amoxicillin-clavulanic acid Alvei, M. Morganii, P. Rettgeri, P. Stuartii, S. -

Prevalence of Proteus Species in Clinical Samples, Antibiotic Sensitivity Pattern and ESBL Production
Int.J.Curr.Microbiol.App.Sci (2013) 2(10): 253-261 ISSN: 2319-7706 Volume 2 Number 10 (2013) pp. 253-261 http://www.ijcmas.com Original Research Article Prevalence of Proteus species in clinical samples, antibiotic sensitivity pattern and ESBL production Jitendra Kumar Pandey1*, Akanksha Narayan2 and Shikhar Tyagi3 Department of Microbiology, MGM Medical College and Hospital, Sector-18, Kamothe, Navi Mumbai- 410209, Maharashtra, India 2Department of Microbiology, Doon (PG) Paramedical College & Hospital, Chakrata Road, Dehradun, Uttarakhand, India 3Department of clinical Research, Jamia Hamdard University, New Delhi, India *Corresponding author A B S T R A C T K e y w o r d s Different Proteus species may vary with the type of infections they cause in both the community and hospital environments. However, in many laboratories in Antimicrobial developing countries, differentiation of the genus Proteus into species is not susceptibility; generally done during bacteriological diagnosis due to high cost and special skills antibiotic involved. This study aimed at determining the prevalence of different Proteus resistance; species in MGM Hospital, Navi Mumbai , their antibiotic resistance pattern and -lactamase. how they relate to patients demographic data. Introduction Proteus species are among the commonly however, bacteriological diagnosis up to implicated pathogens in hospital as well as the identification of species is rare in community acquired infections (Douglas, many laboratories in Ghana due to the cost 2000; Emori, 1993). This pathogen has and special skills involved. There is a diverse mode of transmission, and therefore limited documented information hence can cause infection in different relating to patients demographics and anatomical sites of the body. -

Prevalence of Multidrug (MDR) and Extensively Drug Resistant (XDR) Proteus Species in a Tertiary Care Hospital, India
Int.J.Curr.Microbiol.App.Sci (2014) 3(10) 243-252 ISSN: 2319-7706 Volume 3 Number 10 (2014) pp. 243-252 http://www.ijcmas.com Original Research Article Prevalence of Multidrug (MDR) and Extensively Drug Resistant (XDR) Proteus species in a tertiary care hospital, India Nita Pal*, Nikita Sharma, Rajni Sharma, Saroj Hooja, and Rakesh K Maheshwari Department of Microbiology, SMS Medical College, Jaipur (Rajasthan), India *Corresponding author A B S T R A C T Different Proteus species are among the commonly implicated pathogens in hospital as well as community acquired infections. They can cause infection in different anatomical sites of the body due to different modes of transmission. To determine the prevalence of different K e y w o r d s Proteus species and antibiotic resistance pattern. Isolation and identification of proteus species was done by conventional microbiological procedures. Antibiotic susceptibility testing was performed by Kirby Bauer disc diffusion method. One hundred one Proteus Proteus, species were isolated from 3922 clinical specimens, giving prevalence of 5.38 %. Wound multiple drug isolates were the highest (67.85%) followed by urine (19.64%). Three Proteus species were resistance, isolated; P. mirabilis (62.37 %), P. vulgaris (29.70%) and P. penneri (7.92 %). Males were extensively found to be more vulnerable than females in acquiring Proteus infections. Among the in- drug- patients infection rate was 81.18% and out-patients 18.82 %.All the species were resistant resistance amoxicillin-clavulanic acid, ceftazidime, cefepime, amikacin, gentamycin and netilmycin. Piperacillin-tazobactam and imipenem were the most active antibiotics followed by meropenem. -

Examination of Multidrug-Resistant Enterobacteriaceae Isolated from Salted Cattle Hides and Sheep Skins by M
334 Examination of Multidrug-resistant Enterobacteriaceae Isolated from Salted Cattle Hides and Sheep Skins by M. Birbir,1* K. Ulusoy2 and P. Caglayan1 1Marmara University, Faculty of Arts and Sciences, Department of Biology, Goztepe, Istanbul 34722, Turkey 2Marmara University, Institute of Pure and Applied Science, Goztepe, Istanbul 34722, Turkey Abstract antibiotics. Researchers reported that members of the family Enterobacteriaceae and the genera Pasteurella and Staphylococcus 2 Antibiotic resistance profiles in Enterobacteriaceae isolated from easily can become resistant to certain antibiotics. It has been salted cattle hides and sheep skin samples were examined in this known that genes encoding antibiotic resistance are actually study. Antibiotic resistance profiles of 27 cattle hide and 28 found in every microorganism that produces antibiotics. Genetic sheep skin isolates, obtained from five salted cattle hide and five basis of this resistance may be chromosomal, plasmid or 3 skin samples originating in different countries such as Dubai, both. Antibiotic resistance genes may be transmitted via mobile Turkey, Israel, Australia, Lebanon, U.S.A. and South Africa, genetic elements, transposons and integrons to other 4 were examined by disc diffussion susceptibility method using 24 microorganisms. Inactivation of antibiotic, development of different antimicrobial agents. Seventy percent of the salted hide resistant biochemical pathway, reduced permeability, the isolates and sixty-eight percent of the salted sheep skin isolates multidrug efflux systems which pump antibiotics out of bacterial exhibited resistance to three or more of 24 antimicrobial agents cell, alteration of target and quorum sensing are important 3,5,6 used. Less than 50% of the isolates was resistant to tobramycin resistance mechanisms against various antibiotics. -

Book IJPHRD June 2020.Indb
148 Indian Journal of Public Health Research & Development, June 2020, Vol. 11, No. 6 Clinical Correlation, Isolation, Speciation, and Antibiotic Susceptibility Pattern of Tribe Proteeae from Various Clinical Samples Bavitha C1, Udayalaxmi J*2, Elizabeth G1, Pooja Rao2 1MSc Microbiology student, 2Associate Professor, Microbiology department, Kasturba Medical College, Mangalore, Manipal Academy of Higher Education, Manipal, Karnataka, India Abstract Introduction: Organisms belonging to Tribe Proteeae are usually present as commensals in the environment but can cause urinary tract and wound infections. There is a need for regular studies on sensitivity pattern of various bacteria causing infections to decide a suitable empirical therapy and there are not many studies on Tribe Proteeae. Aim: To clinically correlate, isolate, speciate and determine the antibiotic susceptibility pattern of Tribe Proteeae from various clinical samples. Materials and Method: All samples were cultured on standard culture media as per routine protocol. Isolates were identified by standard biochemical reactions. Antibiotic sensitivity was done by Kirby Bauer’s disk diffusion method as per CLSI guidelines. ESBL production was detected by double disk diffusion method using ceftazidime (30µg) disk and ceftazidime/clavulanic acid (30/10µg) disk. Clinical data was collected from the record section of the hospital. Results: Sixty samples yielded bacteria belonging to Tribe Proteeae. Most of the patients belonged to male gender 46 (76.7%) and >50 years 38 (63.3%) of age. P mirabilis was the predominant isolate 42 (70%). Maximum rate of isolation of Proteus spp was from diabetic foot ulcer 21 (35%). The predisposing conditions were diabetes mellitus, tracheostomy cases and only one case of carcinoma tongue.