Import of Non-Coding Rnas Into Human Mitochondria: a Critical Review and Emerging Approaches
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

Mt-Atp8 Gene in the Conplastic Mouse Strain C57BL/6J-Mtfvb/NJ on the Mitochondrial Function and Consequent Alterations to Metabolic and Immunological Phenotypes
From the Lübeck Institute of Experimental Dermatology of the University of Lübeck Director: Prof. Dr. Saleh M. Ibrahim Interplay of mtDNA, metabolism and microbiota in the pathogenesis of AIBD Dissertation for Fulfillment of Requirements for the Doctoral Degree of the University of Lübeck from the Department of Natural Sciences Submitted by Paul Schilf from Rostock Lübeck, 2016 First referee: Prof. Dr. Saleh M. Ibrahim Second referee: Prof. Dr. Stephan Anemüller Chairman: Prof. Dr. Rainer Duden Date of oral examination: 30.03.2017 Approved for printing: Lübeck, 06.04.2017 Ich versichere, dass ich die Dissertation ohne fremde Hilfe angefertigt und keine anderen als die angegebenen Hilfsmittel verwendet habe. Weder vorher noch gleichzeitig habe ich andernorts einen Zulassungsantrag gestellt oder diese Dissertation vorgelegt. ABSTRACT Mitochondria are critical in the regulation of cellular metabolism and influence signaling processes and inflammatory responses. Mitochondrial DNA mutations and mitochondrial dysfunction are known to cause a wide range of pathological conditions and are associated with various immune diseases. The findings in this work describe the effect of a mutation in the mitochondrially encoded mt-Atp8 gene in the conplastic mouse strain C57BL/6J-mtFVB/NJ on the mitochondrial function and consequent alterations to metabolic and immunological phenotypes. This work provides insights into the mutation-induced cellular adaptations that influence the inflammatory milieu and shape pathological processes, in particular focusing on autoimmune bullous diseases, which have recently been reported to be associated with mtDNA polymorphisms in the human MT-ATP8 gene. The mt-Atp8 mutation diminishes the assembly of the ATP synthase complex into multimers and decreases mitochondrial respiration, affects generation of reactive oxygen species thus leading to a shift in the metabolic balance and reduction in the energy state of the cell as indicated by the ratio ATP to ADP. -

Mitochondrial Complex III Deficiency Associated with a Homozygous Mutation in UQCRQ
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Elsevier - Publisher Connector REPORT Mitochondrial Complex III Deficiency Associated with a Homozygous Mutation in UQCRQ Ortal Barel,1 Zamir Shorer,2 Hagit Flusser,2 Rivka Ofir,1 Ginat Narkis,1 Gal Finer,1 Hanah Shalev,2 Ahmad Nasasra,2 Ann Saada,3 and Ohad S. Birk1,4,* A consanguineous Israeli Bedouin kindred presented with an autosomal-recessive nonlethal phenotype of severe psychomotor retarda- tion and extrapyramidal signs, dystonia, athetosis and ataxia, mild axial hypotonia, and marked global dementia with defects in verbal and expressive communication skills. Metabolic workup was normal except for mildly elevated blood lactate levels. Brain magnetic resonance imaging (MRI) showed increased density in the putamen, with decreased density and size of the caudate and lentiform nuclei. Reduced activity specifically of mitochondrial complex III and variable decrease in complex I activity were evident in muscle biopsies. Homozygosity of affected individuals to UQCRB and to BCSIL, previously associated with isolated complex III deficiency, was ruled out. Genome-wide linkage analysis identified a homozygosity locus of approximately 9 cM on chromosome 5q31 that was further narrowed down to 2.14 cM, harboring 30 genes (logarithm of the odds [LOD] score 8.82 at q ¼ 0). All 30 genes were sequenced, revealing a single missense (p.Ser45Phe) mutation in UQCRQ (encoding ubiquinol-cytochrome c reductase, complex III subunit VII, 9.5 kDa), one of the ten nuclear -

Cartilage-Hair Hypoplasia
Cartilage-hair hypoplasia Description Cartilage-hair hypoplasia is a disorder of bone growth characterized by short stature ( dwarfism) with other skeletal abnormalities; fine, sparse hair (hypotrichosis); and abnormal immune system function (immune deficiency) that can lead to recurrent infections. People with cartilage-hair hypoplasia have unusually short limbs and short stature from birth. They typically have malformations in the cartilage near the ends of the long bones in the arms and legs (metaphyseal chondrodysplasia), which then affects development of the bone itself. Most people with cartilage-hair hypoplasia are unusually flexible in some joints, but they may have difficulty extending their elbows fully. Affected individuals have hair that is lighter in color than that of other family members because the core of each hair, which contains some of the pigment that contributes the hair's color, is missing. The missing core also makes each strand of hair thinner, causing the hair to have a sparse appearance overall. Unusually light-colored skin ( hypopigmentation), malformed nails, and dental abnormalities may also be seen in this disorder. The extent of the immune deficiency in cartilage-hair hypoplasia varies from mild to severe. Affected individuals with the most severe immune problems are considered to have severe combined immunodeficiency (SCID). People with SCID lack virtually all immune protection from bacteria, viruses, and fungi and are prone to repeated and persistent infections that can be very serious or life-threatening. These infections are often caused by "opportunistic" organisms that ordinarily do not cause illness in people with a normal immune system. Most people with cartilage-hair hypoplasia, even those who have milder immune deficiency, experience infections of the respiratory system, ears, and sinuses. -

ARTICLES Functional Mitochondrial Heterogeneity in Heteroplasmic
0031-3998/00/4802-0143 PEDIATRIC RESEARCH Vol. 48, No. 2, 2000 Copyright © 2000 International Pediatric Research Foundation, Inc. Printed in U.S.A. ARTICLES Functional Mitochondrial Heterogeneity in Heteroplasmic Cells Carrying the Mitochondrial DNA Mutation Associated with the MELAS Syndrome (Mitochondrial Encephalopathy, Lactic Acidosis, and Strokelike Episodes) ANNETTE BAKKER, CYRILLE BARTHE´ LE´ MY, PAULE FRACHON, DANIELLE CHATEAU, DAMIEN STERNBERG, JEAN PIERRE MAZAT, AND ANNE LOMBE` S INSERM UR523, Institut de Myologie, 75651 Paris, France [A.B., C.B., P.F., D.C., A.L.]; Biochimie B, Hoˆpital de La Salpeˆtrie`re, 75651 Paris, France [D.S.]; and Universite´ de Bordeaux II, INSERM E99–29, 33076 Bordeaux cedex, France [J.P.M.] ABSTRACT Most mitochondrial DNA (mtDNA) alterations associated wild-type mtDNA, transfer RNA, or protein. Mitochondria in with human disorders are heteroplasmic, i.e. mutant mtDNA these heteroplasmic cells cannot, therefore, be considered a molecules coexist with normal ones within the cell. We ad- single functional unit. (Pediatr Res 48: 143–150, 2000) dressed the possibility of intermitochondrial exchanges through histologic analyses of cybrid clones with increasing proportion of the MELAS (A3243G) mtDNA transfer RNA point mutation. Abbreviations MtDNA-dependent cytochrome c oxidase activity and protein MELAS, mitochondrial myopathy, encephalopathy, lactic composition as well as mitochondrial membrane potential ap- acidosis, and strokelike episodes peared heterogeneous in individual cells from clonal heteroplas- -

Biomarkers of Mitotoxicity After Acute Liver Injury: Further Insights Into the Interpretation of Glutamate Dehydrogenase
Journal of Clinical and Translational Research 10.18053/Jctres/07.202101.005 MINI REVIEW Biomarkers of mitotoxicity after acute liver injury: further insights into the interpretation of glutamate dehydrogenase Mitchell R. McGill1,2* and Hartmut Jaeschke3 1. Department of Environmental Health Sciences, Fay W. Boozman College of Public Health, University of Arkansas for Medical Sciences, 4301 W. Markham St, Little Rock, AR, USA, 72205 2. Department of Pharmacology and Toxicology, College of Medicine, University of Arkansas for Medical Sciences, 4301 W. Markham St., Little Rock, AR, USA, 72205 3. Department of Pharmacology, Toxicology and Therapeutics, University of Kansas Medical Center, 3901 Rainbow Blvd., Kansas City, KS, USA, 66160 *Corresponding author Mitchell R. McGill, PhD Department of Environmental Health Sciences & Department of Pharmacology and Toxicology, University of Arkansas for Medical Sciences, Little Rock, AR Tel: +1 501-526-6696 Email: [email protected] Article information: Received: November 03, 2020 Revised: December 09, 2020 Accepted: December 10, 2020 Journal of Clinical and Translational Research 10.18053/Jctres/07.202101.005 ABSTRACT Background: Acetaminophen (APAP) is a popular analgesic, but overdose causes acute liver injury and sometimes death. Decades of research have revealed that mitochondrial damage is central in the mechanisms of toxicity in rodents, but we know much less about the role of mitochondria in humans. Due to the challenge of procuring liver tissue from APAP overdose patients, non-invasive mechanistic biomarkers are necessary to translate the mechanisms of APAP hepatotoxicity from rodents to patients. It was recently proposed that the mitochondrial matrix enzyme glutamate dehydrogenase (GLDH) can be measured in circulation as a biomarker of mitochondrial damage. -

Yeast Genome Gazetteer P35-65
gazetteer Metabolism 35 tRNA modification mitochondrial transport amino-acid metabolism other tRNA-transcription activities vesicular transport (Golgi network, etc.) nitrogen and sulphur metabolism mRNA synthesis peroxisomal transport nucleotide metabolism mRNA processing (splicing) vacuolar transport phosphate metabolism mRNA processing (5’-end, 3’-end processing extracellular transport carbohydrate metabolism and mRNA degradation) cellular import lipid, fatty-acid and sterol metabolism other mRNA-transcription activities other intracellular-transport activities biosynthesis of vitamins, cofactors and RNA transport prosthetic groups other transcription activities Cellular organization and biogenesis 54 ionic homeostasis organization and biogenesis of cell wall and Protein synthesis 48 plasma membrane Energy 40 ribosomal proteins organization and biogenesis of glycolysis translation (initiation,elongation and cytoskeleton gluconeogenesis termination) organization and biogenesis of endoplasmic pentose-phosphate pathway translational control reticulum and Golgi tricarboxylic-acid pathway tRNA synthetases organization and biogenesis of chromosome respiration other protein-synthesis activities structure fermentation mitochondrial organization and biogenesis metabolism of energy reserves (glycogen Protein destination 49 peroxisomal organization and biogenesis and trehalose) protein folding and stabilization endosomal organization and biogenesis other energy-generation activities protein targeting, sorting and translocation vacuolar and lysosomal -
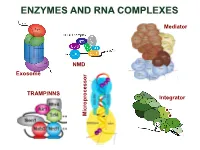
Enzymes and Rna Complexes
ENZYMES AND RNA COMPLEXES Mediator NMD Exosome NMD TRAMP/NNS Integrator Microprocessor RNA PROCESSING and DECAY machinery: RNases Protein Function Characteristics Exonucleases 5’ 3’ Xrn1 cytoplasmic, mRNA degradation processsive Rat1 nuclear, pre-rRNA, sn/snoRNA, pre-mRNA processing and degradation Rrp17/hNol12 nuclear, pre-rRNA processing Exosome 3’ 5’ multisubunit exo/endo complex subunits organized as in bacterial PNPase Rrp44/Dis3 catalytic subunit Exo/PIN domains, processsive Rrp4, Rrp40 pre-rRNA, sn/snoRNA processing, mRNA degradation Rrp41-43, 45-46 participates in NMD, ARE-dependent, non-stop decay Mtr3, Ski4 Mtr4 nuclear helicase cofactor DEAD box Rrp6 (Rrp47) nuclear exonuclease ( Rrp6 BP, cofactor) RNAse D homolog, processsive Ski2,3,7,8 cytoplasmic exosome cofactors. SKI complex helicase, GTPase Other 3’ 5’ Rex1-4 3’-5’ exonucleases, rRNA, snoRNA, tRNA processing RNase D homolog DXO 3’-5’ exonuclease in addition to decapping mtEXO 3’ 5’ mitochondrial degradosome RNA degradation in yeast Suv3/ Dss1 helicase/ 3’-5’ exonuclease DExH box/ RNase II homolog Deadenylation Ccr4/NOT/Pop2 major deadenylase complex (Ccr, Caf, Pop, Not proteins) Ccr4- Mg2+ dependent endonuclease Pan2p/Pan3 additional deadenylases (poliA tail length) RNase D homolog, poly(A) specific nuclease PARN mammalian deadenylase RNase D homolog, poly(A) specific nuclease Endonucleases RNase III -Rnt1 pre-rRNA, sn/snoRNA processing, mRNA degradation dsRNA specific -Dicer, Drosha siRNA/miRNA biogenesis, functions in RNAi PAZ, RNA BD, RNase III domains Ago2 Slicer -

Stouthamer1973
Antonie van Leeuwenhoek 39 (1973) 545-565 545 A theoretical study on the amount of ATP required for synthesis of microbial cell material A. H. STOUTHAMER Biological Laboratory, Free University, de Boelelaan 1087, Amsterdam, the Netherlands STOUTHAMER, A.H. 1973. A theoretical study on the amount of ATP required for synthesis of microbial cell material. Antonie van Leeuwenhoek 39: 545-565. The amount of ATP required for the formation of microbial cells growing under various conditions was calculated. It was assumed that the chemical com position of the cell was the same under all these conditions. The analysis of the chemical composition of microbial cells of Morowitz ( 1968) was taken as a base. It was assumed that 4 moles of ATP are required for the incorporation of one mole of amino acid into protein. The amount of ATP required on account of the instability and frequent regeneration of messenger RNA was calculated from data in the literature pertaining to the relative rates of synthesis of the various classes of RNA molecules in the cell. An estimate is given of the amount of ATP required for transport processes. For this purpose it was assumed that 0.5 mole of ATP is necessary for the uptake of 1 g-ion of potassium or ammo nium, and 1 mole of ATP for the uptake of 1 mole of phosphate, amino acid, acetate, malate etc. The results of the calculations show that from preformed monomers (glucose, amino acids and nucleic acid bases) 31.9 g cells can be formed per g-mole of ATP when acetyl-CoA is formed from glucose. -

A Disease-Linked Lncrna Mutation in Rnase MRP Inhibits Ribosome Synthesis
bioRxiv preprint doi: https://doi.org/10.1101/2021.03.29.437572; this version posted March 29, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. A disease-linked lncRNA mutation in RNase MRP inhibits ribosome synthesis Nic Roberston1, Vadim Shchepachev1, David Wright2, Tomasz W. Turowski1, Christos Spanos1, Aleksandra Helwak1, Rose Zamoyska2, David Tollervey1 1 Wellcome Centre for Cell Biology, University of Edinburgh, Edinburgh, UK 2 Ashworth Laboratories, Institute of Immunology and Infection Research, University of Edinburgh, Edinburgh, UK Keywords: protein-RNA interaction; RNA-binding sites; UV crosslinking; mass spectrometry; genetic disease; Cartilage Hair Hypoplasia; ribosome synthesis; T cell activation Running title: RNase MRP defects cause ribosomopathy Highlights: • Mutations in RMRP lncRNA impair pre-rRNA processing and T cell activation • Patient derived fibroblasts show impaired pre-rRNA processing • Cells with the most common disease-linked mutation have specific processing defects • Cytoplasmic ribosomes and intact RNase MRP complexes are also reduced in these cells 1 bioRxiv preprint doi: https://doi.org/10.1101/2021.03.29.437572; this version posted March 29, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. Abstract Mutations in the human RMRP gene cause Cartilage Hair Hypoplasia (CHH), an autosomal recessive disorder characterized by skeletal abnormalities and impaired T cell activation. -

Tyramine and Amyloid Beta 42: a Toxic Synergy
biomedicines Article Tyramine and Amyloid Beta 42: A Toxic Synergy Sudip Dhakal and Ian Macreadie * School of Science, RMIT University, Bundoora, VIC 3083, Australia; [email protected] * Correspondence: [email protected]; Tel.: +61-3-9925-6627 Received: 5 May 2020; Accepted: 27 May 2020; Published: 30 May 2020 Abstract: Implicated in various diseases including Parkinson’s disease, Huntington’s disease, migraines, schizophrenia and increased blood pressure, tyramine plays a crucial role as a neurotransmitter in the synaptic cleft by reducing serotonergic and dopaminergic signaling through a trace amine-associated receptor (TAAR1). There appear to be no studies investigating a connection of tyramine to Alzheimer’s disease. This study aimed to examine whether tyramine could be involved in AD pathology by using Saccharomyces cerevisiae expressing Aβ42. S. cerevisiae cells producing native Aβ42 were treated with different concentrations of tyramine, and the production of reactive oxygen species (ROS) was evaluated using flow cytometric cell analysis. There was dose-dependent ROS generation in wild-type yeast cells with tyramine. In yeast producing Aβ42, ROS levels generated were significantly higher than in controls, suggesting a synergistic toxicity of Aβ42 and tyramine. The addition of exogenous reduced glutathione (GSH) was found to rescue the cells with increased ROS, indicating depletion of intracellular GSH due to tyramine and Aβ42. Additionally, tyramine inhibited the respiratory growth of yeast cells producing GFP-Aβ42, while there was no growth inhibition when cells were producing GFP. Tyramine was also demonstrated to cause increased mitochondrial DNA damage, resulting in the formation of petite mutants that lack respiratory function. -

A Specialized Processing Body That Is Temporally and Asymmetrically Regulated During the Cell Cycle in Saccharomyces Cerevisiae
JCB: ARTICLE A specialized processing body that is temporally and asymmetrically regulated during the cell cycle in Saccharomyces cerevisiae Tina Gill, Jason Aulds, and Mark E. Schmitt Department of Biochemistry and Molecular Biology, State University of New York Upstate Medical University, Syracuse, NY 13210 Nase mitochondrial RNA processing (MRP) is an this spot was asymmetrically found in daughter cells, essential ribonucleoprotein endoribonuclease that where the RNase MRP substrate, CLB2 mRNA, localizes. R functions in the degradation of specifi c mRNAs in- Both the mitotic exit network and fourteen early ana- volved in cell cycle regulation. We have investigated phase release pathways are nonessential but important where this processing event occurs and how it is regu- for the temporal changes in localization. Asymmetric lo- lated. As expected, results demonstrate that RNase MRP calization was found to be dependent on the locasome. is predominantly localized in the nucleolus, where it The evidence suggests that these spots are specialized processes ribosomal RNAs. However, after the initiation processing bodies for the degradation of transcripts that of mitosis, RNase MRP localizes throughout the entire are cell cycle regulated and daughter cell localized. We nucleus and in a single discrete cytoplasmic spot that have called these TAM bodies for temporal asymmetric persists until the completion of telophase. Furthermore, MRP bodies. Introduction RNase mitochondrial RNA processing (MRP) is an essential ri- 27SA preribosomal RNA at the A3 site, forming the 5.8S(s) bonucleoprotein endoribonuclease that cleaves RNA substrates ribosomal RNA (rRNA; Schmitt and Clayton, 1993; Lygerou in a site-specifi c manner and is highly conserved in eukaryotes et al., 1996). -

Progressive Increase in Mtdna 3243A>G Heteroplasmy Causes
Progressive increase in mtDNA 3243A>G PNAS PLUS heteroplasmy causes abrupt transcriptional reprogramming Martin Picarda, Jiangwen Zhangb, Saege Hancockc, Olga Derbenevaa, Ryan Golhard, Pawel Golike, Sean O’Hearnf, Shawn Levyg, Prasanth Potluria, Maria Lvovaa, Antonio Davilaa, Chun Shi Lina, Juan Carlos Perinh, Eric F. Rappaporth, Hakon Hakonarsonc, Ian A. Trouncei, Vincent Procaccioj, and Douglas C. Wallacea,1 aCenter for Mitochondrial and Epigenomic Medicine, Children’s Hospital of Philadelphia and the Department of Pathology and Laboratory Medicine, University of Pennsylvania, Philadelphia, PA 19104; bSchool of Biological Sciences, The University of Hong Kong, Hong Kong, People’s Republic of China; cTrovagene, San Diego, CA 92130; dCenter for Applied Genomics, Division of Genetics, Department of Pediatrics, and hNucleic Acid/Protein Research Core Facility, Children’s Hospital of Philadelphia, Philadelphia, PA 19104; eInstitute of Genetics and Biotechnology, Warsaw University, 00-927, Warsaw, Poland; fMorton Mower Central Research Laboratory, Sinai Hospital of Baltimore, Baltimore, MD 21215; gGenomics Sevices Laboratory, HudsonAlpha Institute for Biotechnology, Huntsville, AL 35806; iCentre for Eye Research Australia, Royal Victorian Eye and Ear Hospital, East Melbourne, VIC 3002, Australia; and jDepartment of Biochemistry and Genetics, National Center for Neurodegenerative and Mitochondrial Diseases, Centre Hospitalier Universitaire d’Angers, 49933 Angers, France Contributed by Douglas C. Wallace, August 1, 2014 (sent for review May