Histone Variant H2A.J Accumulates in Senescent Cells and Promotes
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

Histone Isoform H2A1H Promotes Attainment of Distinct Physiological
Bhattacharya et al. Epigenetics & Chromatin (2017) 10:48 DOI 10.1186/s13072-017-0155-z Epigenetics & Chromatin RESEARCH Open Access Histone isoform H2A1H promotes attainment of distinct physiological states by altering chromatin dynamics Saikat Bhattacharya1,4,6, Divya Reddy1,4, Vinod Jani5†, Nikhil Gadewal3†, Sanket Shah1,4, Raja Reddy2,4, Kakoli Bose2,4, Uddhavesh Sonavane5, Rajendra Joshi5 and Sanjay Gupta1,4* Abstract Background: The distinct functional efects of the replication-dependent histone H2A isoforms have been dem- onstrated; however, the mechanistic basis of the non-redundancy remains unclear. Here, we have investigated the specifc functional contribution of the histone H2A isoform H2A1H, which difers from another isoform H2A2A3 in the identity of only three amino acids. Results: H2A1H exhibits varied expression levels in diferent normal tissues and human cancer cell lines (H2A1C in humans). It also promotes cell proliferation in a context-dependent manner when exogenously overexpressed. To uncover the molecular basis of the non-redundancy, equilibrium unfolding of recombinant H2A1H-H2B dimer was performed. We found that the M51L alteration at the H2A–H2B dimer interface decreases the temperature of melting of H2A1H-H2B by ~ 3 °C as compared to the H2A2A3-H2B dimer. This diference in the dimer stability is also refected in the chromatin dynamics as H2A1H-containing nucleosomes are more stable owing to M51L and K99R substitu- tions. Molecular dynamic simulations suggest that these substitutions increase the number of hydrogen bonds and hydrophobic interactions of H2A1H, enabling it to form more stable nucleosomes. Conclusion: We show that the M51L and K99R substitutions, besides altering the stability of histone–histone and histone–DNA complexes, have the most prominent efect on cell proliferation, suggesting that the nucleosome sta- bility is intimately linked with the physiological efects observed. -

Environmental Influences on Endothelial Gene Expression
ENDOTHELIAL CELL GENE EXPRESSION John Matthew Jeff Herbert Supervisors: Prof. Roy Bicknell and Dr. Victoria Heath PhD thesis University of Birmingham August 2012 University of Birmingham Research Archive e-theses repository This unpublished thesis/dissertation is copyright of the author and/or third parties. The intellectual property rights of the author or third parties in respect of this work are as defined by The Copyright Designs and Patents Act 1988 or as modified by any successor legislation. Any use made of information contained in this thesis/dissertation must be in accordance with that legislation and must be properly acknowledged. Further distribution or reproduction in any format is prohibited without the permission of the copyright holder. ABSTRACT Tumour angiogenesis is a vital process in the pathology of tumour development and metastasis. Targeting markers of tumour endothelium provide a means of targeted destruction of a tumours oxygen and nutrient supply via destruction of tumour vasculature, which in turn ultimately leads to beneficial consequences to patients. Although current anti -angiogenic and vascular targeting strategies help patients, more potently in combination with chemo therapy, there is still a need for more tumour endothelial marker discoveries as current treatments have cardiovascular and other side effects. For the first time, the analyses of in-vivo biotinylation of an embryonic system is performed to obtain putative vascular targets. Also for the first time, deep sequencing is applied to freshly isolated tumour and normal endothelial cells from lung, colon and bladder tissues for the identification of pan-vascular-targets. Integration of the proteomic, deep sequencing, public cDNA libraries and microarrays, delivers 5,892 putative vascular targets to the science community. -

Repertoire of Morphable Proteins in an Organism
Repertoire of morphable proteins in an organism Keisuke Izumi, Eitaro Saho, Ayuka Kutomi, Fumiaki Tomoike and Tetsuji Okada Department of Life Science, Gakushuin University, Tokyo, Japan ABSTRACT All living organisms have evolved to contain a set of proteins with variable physical and chemical properties. Efforts in the field of structural biology have contributed to uncovering the shape and the variability of each component. However, quantification of the variability has been performed mostly by multiple pair-wise comparisons. A set of experimental coordinates for a given protein can be used to define the “morphness/unmorphness”. To understand the evolved repertoire in an organism, here we show the results of global analysis of more than a thousand Escherichia coli proteins, by the recently introduced method, distance scoring analysis (DSA). By collecting a new index “UnMorphness Factor” (UMF), proposed in this study and determined from DSA for each of the proteins, the lowest and the highest boundaries of the experimentally observable structural variation are comprehensibly defined. The distribution plot of UMFs obtained for E. coli represents the first view of a substantial fraction of non-redundant proteome set of an organism, demonstrating how rigid and flexible components are balanced. The present analysis extends to evaluate the growing data from single particle cryo-electron microscopy, providing valuable information on effective interpretation to structural changes of proteins and the supramolecular complexes. Subjects Biochemistry, Bioinformatics, Biophysics, Molecular Biology Keywords Protein, Structure, Crystallography, cryo-EM, E. coli, Human, PDB, Coordinates Submitted 21 October 2019 Accepted 20 January 2020 INTRODUCTION Published 11 February 2020 Self-replicating (living) species are defined by the presence of a genome that is used to Corresponding author Tetsuji Okada, produce a set of proteins and RNAs. -

Use of Genomic Tools to Discover the Cause of Champagne Dilution Coat Color in Horses and to Map the Genetic Cause of Extreme Lordosis in American Saddlebred Horses
University of Kentucky UKnowledge Theses and Dissertations--Veterinary Science Veterinary Science 2014 USE OF GENOMIC TOOLS TO DISCOVER THE CAUSE OF CHAMPAGNE DILUTION COAT COLOR IN HORSES AND TO MAP THE GENETIC CAUSE OF EXTREME LORDOSIS IN AMERICAN SADDLEBRED HORSES Deborah G. Cook University of Kentucky, [email protected] Right click to open a feedback form in a new tab to let us know how this document benefits ou.y Recommended Citation Cook, Deborah G., "USE OF GENOMIC TOOLS TO DISCOVER THE CAUSE OF CHAMPAGNE DILUTION COAT COLOR IN HORSES AND TO MAP THE GENETIC CAUSE OF EXTREME LORDOSIS IN AMERICAN SADDLEBRED HORSES" (2014). Theses and Dissertations--Veterinary Science. 15. https://uknowledge.uky.edu/gluck_etds/15 This Doctoral Dissertation is brought to you for free and open access by the Veterinary Science at UKnowledge. It has been accepted for inclusion in Theses and Dissertations--Veterinary Science by an authorized administrator of UKnowledge. For more information, please contact [email protected]. STUDENT AGREEMENT: I represent that my thesis or dissertation and abstract are my original work. Proper attribution has been given to all outside sources. I understand that I am solely responsible for obtaining any needed copyright permissions. I have obtained needed written permission statement(s) from the owner(s) of each third-party copyrighted matter to be included in my work, allowing electronic distribution (if such use is not permitted by the fair use doctrine) which will be submitted to UKnowledge as Additional File. I hereby grant to The University of Kentucky and its agents the irrevocable, non-exclusive, and royalty-free license to archive and make accessible my work in whole or in part in all forms of media, now or hereafter known. -

Differences for Ectopic Versus Eutopic Cells
556 RBMO VOLUME 39 ISSUE 4 2019 ARTICLE Chemosensitivity and chemoresistance in endometriosis – differences for ectopic versus eutopic cells BIOGRAPHY Andres Salumets is Professor of Reproductive Medicine at the University of Tartu, and Scientific Head at the Competence Centre on Health Technologies, Tartu, Estonia. He has been involved in assisted reproduction for 20 years, first as an embryologist and later as a researcher. His major interests are endometriosis, endometrial biology and implantation. Darja Lavogina1,2,*, Külli Samuel1, Arina Lavrits1,3, Alvin Meltsov1, Deniss Sõritsa1,4,5, Ülle Kadastik6, Maire Peters1,4, Ago Rinken2, Andres Salumets1,4,7, 8 KEY MESSAGE Akt/PKB inhibitor GSK690693, CK2 inhibitor ARC-775, MAPK pathway inhibitor sorafenib, proteasome inhibitor bortezomib, and microtubule-depolymerizing toxin MMAE showed higher cytotoxicity in eutopic cells. In contrast, 10 µmol/l of the anthracycline toxin doxorubicin caused cellular death in ectopic cells more effectively than in eutopic cells, underlining the potential of doxorubicin in endometriosis research. ABSTRACT Research question: Endometriosis is a common gynaecological disease defined by the presence of endometrium-like tissue outside the uterus. This complex disease, often accompanied by severe pain and infertility, causes a significant medical and socioeconomic burden; hence, novel strategies are being sought for the treatment of endometriosis. Here, we set out to explore the cytotoxic effects of a panel of compounds to find toxins with different efficiency in eutopic versus ectopic cells, thus highlighting alterations in the corresponding molecular pathways. Design: The effect on cellular viability of 14 compounds was established in a cohort of paired eutopic and ectopic endometrial stromal cell samples from 11 patients. -

HIST1H2AC Human Shrna Plasmid Kit (Locus ID 8334) Product Data
OriGene Technologies, Inc. 9620 Medical Center Drive, Ste 200 Rockville, MD 20850, US Phone: +1-888-267-4436 [email protected] EU: [email protected] CN: [email protected] Product datasheet for TL312444 HIST1H2AC Human shRNA Plasmid Kit (Locus ID 8334) Product data: Product Type: shRNA Plasmids Product Name: HIST1H2AC Human shRNA Plasmid Kit (Locus ID 8334) Locus ID: 8334 Synonyms: dJ221C16.4; H2A/l; H2AFL; HIST1H2AC Vector: pGFP-C-shLenti (TR30023) Format: Lentiviral plasmids Components: HIST1H2AC - Human, 4 unique 29mer shRNA constructs in lentiviral GFP vector(Gene ID = 8334). 5µg purified plasmid DNA per construct Non-effective 29-mer scrambled shRNA cassette in pGFP-C-shLenti Vector, TR30021, included for free. RefSeq: NM_003512, NM_003512.1, NM_003512.2, NM_003512.3, BC085010, BC017379, BC050602, NM_003512.4 Summary: Histones are basic nuclear proteins that are responsible for the nucleosome structure of the chromosomal fiber in eukaryotes. Two molecules of each of the four core histones (H2A, H2B, H3, and H4) form an octamer, around which approximately 146 bp of DNA is wrapped in repeating units, called nucleosomes. The linker histone, H1, interacts with linker DNA between nucleosomes and functions in the compaction of chromatin into higher order structures. This gene is intronless and encodes a replication-dependent histone that is a member of the histone H2A family. Transcripts from this gene lack polyA tails but instead contain a palindromic termination element. This gene is found in the large histone gene cluster on chromosome 6. [provided by RefSeq, Aug 2015] shRNA Design: These shRNA constructs were designed against multiple splice variants at this gene locus. -

Supplemental Information
Supplemental information Dissection of the genomic structure of the miR-183/96/182 gene. Previously, we showed that the miR-183/96/182 cluster is an intergenic miRNA cluster, located in a ~60-kb interval between the genes encoding nuclear respiratory factor-1 (Nrf1) and ubiquitin-conjugating enzyme E2H (Ube2h) on mouse chr6qA3.3 (1). To start to uncover the genomic structure of the miR- 183/96/182 gene, we first studied genomic features around miR-183/96/182 in the UCSC genome browser (http://genome.UCSC.edu/), and identified two CpG islands 3.4-6.5 kb 5’ of pre-miR-183, the most 5’ miRNA of the cluster (Fig. 1A; Fig. S1 and Seq. S1). A cDNA clone, AK044220, located at 3.2-4.6 kb 5’ to pre-miR-183, encompasses the second CpG island (Fig. 1A; Fig. S1). We hypothesized that this cDNA clone was derived from 5’ exon(s) of the primary transcript of the miR-183/96/182 gene, as CpG islands are often associated with promoters (2). Supporting this hypothesis, multiple expressed sequences detected by gene-trap clones, including clone D016D06 (3, 4), were co-localized with the cDNA clone AK044220 (Fig. 1A; Fig. S1). Clone D016D06, deposited by the German GeneTrap Consortium (GGTC) (http://tikus.gsf.de) (3, 4), was derived from insertion of a retroviral construct, rFlpROSAβgeo in 129S2 ES cells (Fig. 1A and C). The rFlpROSAβgeo construct carries a promoterless reporter gene, the β−geo cassette - an in-frame fusion of the β-galactosidase and neomycin resistance (Neor) gene (5), with a splicing acceptor (SA) immediately upstream, and a polyA signal downstream of the β−geo cassette (Fig. -

Folate Cycle Enzyme MTHFD1L Confers Metabolic Advantages in Hepatocellular Carcinoma
RESEARCH ARTICLE The Journal of Clinical Investigation Folate cycle enzyme MTHFD1L confers metabolic advantages in hepatocellular carcinoma Derek Lee,1 Iris Ming-Jing Xu,1 David Kung-Chun Chiu,1 Robin Kit-Ho Lai,1 Aki Pui-Wah Tse,1 Lynna Lan Li,1 Cheuk-Ting Law,1 Felice Ho-Ching Tsang,1 Larry Lai Wei,1 Cerise Yuen-Ki Chan,1 Chun-Ming Wong,1,2 Irene Oi-Lin Ng,1,2 and Carmen Chak-Lui Wong1,2 1Department of Pathology and 2State Key Laboratory for Liver Research, The University of Hong Kong, Hong Kong, China. Cancer cells preferentially utilize glucose and glutamine, which provide macromolecules and antioxidants that sustain rapid cell division. Metabolic reprogramming in cancer drives an increased glycolytic rate that supports maximal production of these nutrients. The folate cycle, through transfer of a carbon unit between tetrahydrofolate and its derivatives in the cytoplasmic and mitochondrial compartments, produces other metabolites that are essential for cell growth, including nucleotides, methionine, and the antioxidant NADPH. Here, using hepatocellular carcinoma (HCC) as a cancer model, we have observed a reduction in growth rate upon withdrawal of folate. We found that an enzyme in the folate cycle, methylenetetrahydrofolate dehydrogenase 1–like (MTHFD1L), plays an essential role in support of cancer growth. We determined that MTHFD1L is transcriptionally activated by NRF2, a master regulator of redox homeostasis. Our observations further suggest that MTHFD1L contributes to the production and accumulation of NADPH to levels that are sufficient to combat oxidative stress in cancer cells. The elevation of oxidative stress through MTHFD1L knockdown or the use of methotrexate, an antifolate drug, sensitizes cancer cells to sorafenib, a targeted therapy for HCC. -

Supplementary Table S4. FGA Co-Expressed Gene List in LUAD
Supplementary Table S4. FGA co-expressed gene list in LUAD tumors Symbol R Locus Description FGG 0.919 4q28 fibrinogen gamma chain FGL1 0.635 8p22 fibrinogen-like 1 SLC7A2 0.536 8p22 solute carrier family 7 (cationic amino acid transporter, y+ system), member 2 DUSP4 0.521 8p12-p11 dual specificity phosphatase 4 HAL 0.51 12q22-q24.1histidine ammonia-lyase PDE4D 0.499 5q12 phosphodiesterase 4D, cAMP-specific FURIN 0.497 15q26.1 furin (paired basic amino acid cleaving enzyme) CPS1 0.49 2q35 carbamoyl-phosphate synthase 1, mitochondrial TESC 0.478 12q24.22 tescalcin INHA 0.465 2q35 inhibin, alpha S100P 0.461 4p16 S100 calcium binding protein P VPS37A 0.447 8p22 vacuolar protein sorting 37 homolog A (S. cerevisiae) SLC16A14 0.447 2q36.3 solute carrier family 16, member 14 PPARGC1A 0.443 4p15.1 peroxisome proliferator-activated receptor gamma, coactivator 1 alpha SIK1 0.435 21q22.3 salt-inducible kinase 1 IRS2 0.434 13q34 insulin receptor substrate 2 RND1 0.433 12q12 Rho family GTPase 1 HGD 0.433 3q13.33 homogentisate 1,2-dioxygenase PTP4A1 0.432 6q12 protein tyrosine phosphatase type IVA, member 1 C8orf4 0.428 8p11.2 chromosome 8 open reading frame 4 DDC 0.427 7p12.2 dopa decarboxylase (aromatic L-amino acid decarboxylase) TACC2 0.427 10q26 transforming, acidic coiled-coil containing protein 2 MUC13 0.422 3q21.2 mucin 13, cell surface associated C5 0.412 9q33-q34 complement component 5 NR4A2 0.412 2q22-q23 nuclear receptor subfamily 4, group A, member 2 EYS 0.411 6q12 eyes shut homolog (Drosophila) GPX2 0.406 14q24.1 glutathione peroxidase -
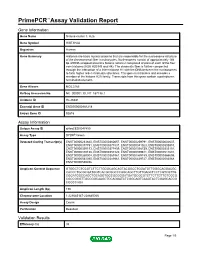
Primepcr™Assay Validation Report
PrimePCR™Assay Validation Report Gene Information Gene Name histone cluster 3, H2a Gene Symbol HIST3H2A Organism Human Gene Summary Histones are basic nuclear proteins that are responsible for the nucleosome structure of the chromosomal fiber in eukaryotes. Nucleosomes consist of approximately 146 bp of DNA wrapped around a histone octamer composed of pairs of each of the four core histones (H2A H2B H3 and H4). The chromatin fiber is further compacted through the interaction of a linker histone H1 with the DNA between the nucleosomes to form higher order chromatin structures. This gene is intronless and encodes a member of the histone H2A family. Transcripts from this gene contain a palindromic termination element. Gene Aliases MGC3165 RefSeq Accession No. NC_000001.10, NT_167186.1 UniGene ID Hs.26331 Ensembl Gene ID ENSG00000181218 Entrez Gene ID 92815 Assay Information Unique Assay ID qHsaCED0047450 Assay Type SYBR® Green Detected Coding Transcript(s) ENST00000233840, ENST00000360657, ENST00000259791, ENST00000602637, ENST00000377791, ENST00000377831, ENST00000341023, ENST00000303910, ENST00000359193, ENST00000377459, ENST00000358739, ENST00000333151, ENST00000330180, ENST00000357320, ENST00000359611, ENST00000511601, ENST00000260008, ENST00000369161, ENST00000369159, ENST00000366695, ENST00000543095, ENST00000389961, ENST00000439727, ENST00000583968, ENST00000580456 Amplicon Context Sequence GTGGCTCTCCGTCTTCTTGGGCAGCAGTACGGCCTGGATGTTGGGCAGGACGC CACCCTGCGCGATGGTCACGCGGCCCAGCAGCTTGTTGAGCTCCTCGTCGTTG CGGATGGCCAGCTGCAGGTGGCGCGGGATGATGCGCGTCTTCTTGTTGTCGCG -

A Mutation in Histone H2B Represents a New Class of Oncogenic Driver
Author Manuscript Published OnlineFirst on July 23, 2019; DOI: 10.1158/2159-8290.CD-19-0393 Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. A Mutation in Histone H2B Represents A New Class Of Oncogenic Driver Richard L. Bennett1, Aditya Bele1, Eliza C. Small2, Christine M. Will2, Behnam Nabet3, Jon A. Oyer2, Xiaoxiao Huang1,9, Rajarshi P. Ghosh4, Adrian T. Grzybowski5, Tao Yu6, Qiao Zhang7, Alberto Riva8, Tanmay P. Lele7, George C. Schatz9, Neil L. Kelleher9 Alexander J. Ruthenburg5, Jan Liphardt4 and Jonathan D. Licht1 * 1 Division of Hematology/Oncology, University of Florida Health Cancer Center, Gainesville, FL 2 Division of Hematology/Oncology, Northwestern University 3 Department of Cancer Biology, Dana Farber Cancer Institute and Department of Biological Chemistry and Molecular Pharmacology, Harvard Medical School 4 Department of Bioengineering, Stanford University 5 Department of Molecular Genetics and Cell Biology, The University of Chicago 6 Department of Chemistry, Tennessee Technological University 7 Department of Chemical Engineering, University of Florida 8 Bioinformatics Core, Interdisciplinary Center for Biotechnology Research, University of Florida 9 Department of Chemistry, Northwestern University, Evanston IL 60208 Running title: Histone mutations in cancer *Corresponding Author: Jonathan D. Licht, MD The University of Florida Health Cancer Center Cancer and Genetics Research Complex, Suite 145 2033 Mowry Road Gainesville, FL 32610 352-273-8143 [email protected] Disclosures: The authors have no conflicts of interest to declare Downloaded from cancerdiscovery.aacrjournals.org on September 27, 2021. © 2019 American Association for Cancer Research. Author Manuscript Published OnlineFirst on July 23, 2019; DOI: 10.1158/2159-8290.CD-19-0393 Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. -

A Discovery Resource of Rare Copy Number Variations in Individuals with Autism Spectrum Disorder
INVESTIGATION A Discovery Resource of Rare Copy Number Variations in Individuals with Autism Spectrum Disorder Aparna Prasad,* Daniele Merico,* Bhooma Thiruvahindrapuram,* John Wei,* Anath C. Lionel,*,† Daisuke Sato,* Jessica Rickaby,* Chao Lu,* Peter Szatmari,‡ Wendy Roberts,§ Bridget A. Fernandez,** Christian R. Marshall,*,†† Eli Hatchwell,‡‡ Peggy S. Eis,‡‡ and Stephen W. Scherer*,†,††,1 *The Centre for Applied Genomics, Program in Genetics and Genome Biology, The Hospital for Sick Children, Toronto M5G 1L7, Canada, †Department of Molecular Genetics, University of Toronto, Toronto M5G 1L7, Canada, ‡Offord Centre for Child Studies, Department of Psychiatry and Behavioural Neurosciences, McMaster University, Hamilton L8P 3B6, § Canada, Autism Research Unit, The Hospital for Sick Children, Toronto M5G 1X8, Canada, **Disciplines of Genetics and Medicine, Memorial University of Newfoundland, St. John’s, Newfoundland A1B 3V6, Canada, ††McLaughlin Centre, University of Toronto, Toronto M5G 1L7, Canada, and ‡‡Population Diagnostics, Inc., Melville, New York 11747 ABSTRACT The identification of rare inherited and de novo copy number variations (CNVs) in human KEYWORDS subjects has proven a productive approach to highlight risk genes for autism spectrum disorder (ASD). A rare variants variety of microarrays are available to detect CNVs, including single-nucleotide polymorphism (SNP) arrays gene copy and comparative genomic hybridization (CGH) arrays. Here, we examine a cohort of 696 unrelated ASD number cases using a high-resolution one-million feature CGH microarray, the majority of which were previously chromosomal genotyped with SNP arrays. Our objective was to discover new CNVs in ASD cases that were not detected abnormalities by SNP microarray analysis and to delineate novel ASD risk loci via combined analysis of CGH and SNP array cytogenetics data sets on the ASD cohort and CGH data on an additional 1000 control samples.