Determining the Differential Roles of the Dock Family Of
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

IDF Patient & Family Handbook
Immune Deficiency Foundation Patient & Family Handbook for Primary Immunodeficiency Diseases This book contains general medical information which cannot be applied safely to any individual case. Medical knowledge and practice can change rapidly. Therefore, this book should not be used as a substitute for professional medical advice. FIFTH EDITION COPYRIGHT 1987, 1993, 2001, 2007, 2013 IMMUNE DEFICIENCY FOUNDATION Copyright 2013 by Immune Deficiency Foundation, USA. REPRINT 2015 Readers may redistribute this article to other individuals for non-commercial use, provided that the text, html codes, and this notice remain intact and unaltered in any way. The Immune Deficiency Foundation Patient & Family Handbook may not be resold, reprinted or redistributed for compensation of any kind without prior written permission from the Immune Deficiency Foundation. If you have any questions about permission, please contact: Immune Deficiency Foundation, 110 West Road, Suite 300, Towson, MD 21204, USA; or by telephone at 800-296-4433. Immune Deficiency Foundation Patient & Family Handbook for Primary Immunodeficency Diseases 5th Edition This publication has been made possible through a generous grant from Baxalta Incorporated Immune Deficiency Foundation 110 West Road, Suite 300 Towson, MD 21204 800-296-4433 www.primaryimmune.org [email protected] EDITORS R. Michael Blaese, MD, Executive Editor Francisco A. Bonilla, MD, PhD Immune Deficiency Foundation Boston Children’s Hospital Towson, MD Boston, MA E. Richard Stiehm, MD M. Elizabeth Younger, CPNP, PhD University of California Los Angeles Johns Hopkins Los Angeles, CA Baltimore, MD CONTRIBUTORS Mark Ballow, MD Joseph Bellanti, MD R. Michael Blaese, MD William Blouin, MSN, ARNP, CPNP State University of New York Georgetown University Hospital Immune Deficiency Foundation Miami Children’s Hospital Buffalo, NY Washington, DC Towson, MD Miami, FL Francisco A. -

Dock3 Protects Myelin in the Cuprizone Model for Demyelination
Citation: Cell Death and Disease (2014) 5, e1395; doi:10.1038/cddis.2014.357 OPEN & 2014 Macmillan Publishers Limited All rights reserved 2041-4889/14 www.nature.com/cddis Dock3 protects myelin in the cuprizone model for demyelination K Namekata1, A Kimura1, C Harada1, H Yoshida2, Y Matsumoto1 and T Harada*,1,2 Dedicator of cytokinesis 3 (Dock3) belongs to an atypical family of the guanine nucleotide exchange factors. It is predominantly expressed in the neural tissues and causes cellular morphological changes by activating the small GTPase Rac1. We previously reported that Dock3 overexpression protects retinal ganglion cells from excitotoxic cell death. Oligodendrocytes are the myelinating cells of axons in the central nervous system and these cells are damaged in demyelinating disorders including multiple sclerosis (MS) and optic neuritis. In this study, we examined if Dock3 is expressed in oligodendrocytes and if increasing Dock3 signals can suppress demyelination in a cuprizone-induced demyelination model, an animal model of MS. We demonstrate that Dock3 is expressed in oligodendrocytes and Dock3 overexpression protects myelin in the corpus callosum following cuprizone treatment. Furthermore, we show that cuprizone demyelinates optic nerves and the extent of demyelination is ameliorated in mice overexpressing Dock3. Cuprizone treatment impairs visual function, which was demonstrated by multifocal electroretinograms, an established non-invasive method, and Dock3 overexpression prevented this effect. In mice overexpressing Dock3, Erk activation is increased, suggesting this may at least partly explain the observed protective effects. Our findings suggest that Dock3 may be a therapeutic target for demyelinating disorders including optic neuritis. Cell Death and Disease (2014) 5, e1395; doi:10.1038/cddis.2014.357; published online 28 August 2014 Dedicator of cytokinesis 3 (Dock3), an atypical member of the will restore the visual function. -

Structure of the Dock2âˆ'elmo1 Complex Provides Insights Into
ARTICLE https://doi.org/10.1038/s41467-020-17271-9 OPEN Structure of the DOCK2−ELMO1 complex provides insights into regulation of the auto-inhibited state Leifu Chang1,7, Jing Yang1, Chang Hwa Jo 2, Andreas Boland 1,8, Ziguo Zhang 1, Stephen H. McLaughlin 1, Afnan Abu-Thuraia 3, Ryan C. Killoran2, Matthew J. Smith 2,4,9, Jean-Francois Côté 3,5,6,9 & ✉ David Barford 1 DOCK (dedicator of cytokinesis) proteins are multidomain guanine nucleotide exchange 1234567890():,; factors (GEFs) for RHO GTPases that regulate intracellular actin dynamics. DOCK proteins share catalytic (DOCKDHR2) and membrane-associated (DOCKDHR1) domains. The structurally-related DOCK1 and DOCK2 GEFs are specific for RAC, and require ELMO (engulfment and cell motility) proteins for function. The N-terminal RAS-binding domain (RBD) of ELMO (ELMORBD) interacts with RHOG to modulate DOCK1/2 activity. Here, we determine the cryo-EM structures of DOCK2−ELMO1 alone, and as a ternary complex with RAC1, together with the crystal structure of a RHOG−ELMO2RBD complex. The binary DOCK2−ELMO1 complex adopts a closed, auto-inhibited conformation. Relief of auto- inhibition to an active, open state, due to a conformational change of the ELMO1 subunit, exposes binding sites for RAC1 on DOCK2DHR2, and RHOG and BAI GPCRs on ELMO1. Our structure explains how up-stream effectors, including DOCK2 and ELMO1 phosphorylation, destabilise the auto-inhibited state to promote an active GEF. 1 MRC Laboratory of Molecular Biology, Cambridge CB2 0QH, UK. 2 Institute for Research in Immunology and Cancer, Université de Montréal, Montréal, Québec H3T 1J4, Canada. 3 Montreal Institute of Clinical Research (IRCM), Montréal, QC H2W 1R7, Canada. -
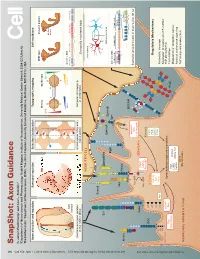
Snapshot: Axon Guidance Pasterkamp R
494 1 Cell Cell ??? SnapShot: Axon Guidance 153 SnapShot: XXXXXXXXXXXXXXXXXXXXXXXXXX 1 2 , ??MONTH?? ??DATE??, 200? ©200? Elsevier Inc. 200?©200? ElsevierInc. , ??MONTH?? ??DATE??, DOI R. Jeroen Pasterkamp and Alex L. Kolodkin , April11, 2013©2013Elsevier Inc. DOI http://dx.doi.org/10.1016/j.cell.2013.03.031 AUTHOR XXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXX 1 AFFILIATIONDepartment of XXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXX Neuroscience and Pharmacology, Rudolf Magnus Institute of Neuroscience, University Medical Center Utrecht, 3584 CG Utrecht, The Netherlands; 2Department of Neuroscience, HHMI, The Johns Hopkins University School of Medicine, Baltimore, MD 21212, USA Axon attraction and repulsion Surround repulsion Selective fasciculation Topographic mapping Self-avoidance Wild-type Dscam1 mutant Sema3A A Retina SC/Tectum EphB EphrinB P D D Mutant T A neuron N P A V V Genomic DNA P EphA EphrinA Slit Exon 4 (12) Exon 6 (48) Exon 9 (33) Exon 17 (2) Netrin Commissural axon guidance Surround repulsion of peripheral Grasshopper CNS axon Retinotectal mapping at the CNS midline nerves in vertebrates fasciculation in vertebrates Drosophila mushroom body XXXXXXXXX NEURITE/CELL Isoneuronal Sema3 Slit Sema1/4-6 Heteroneuronal EphrinA EphrinB FasII Eph Genomic DNA Pcdh-α (14) Pcdh-β (22) Pcdh-γ (22) Variable Con Variable Con Nrp Plexin ** *** Netrin LAMELLIPODIA Con DSCAM ephexin Starburst amacrine cells in mammalian retina Ras-GTP Vav See online version for legend and references. α-chimaerin GEFs/GAPs Robo FARP Ras-GDP LARG RhoGEF Kinases DCC cc0 GTPases PKA cc1 FAK Regulatory Mechanisms See online versionfor??????. Cdc42 GSK3 cc2 Rac PI3K P1 Rho P2 cc3 Abl Proteolytic cleavage P3 Regulation of expression (TF, miRNA, FILOPODIA srGAP Cytoskeleton regulatory proteins multiple isoforms) Sos Trio Pcdh Cis inhibition DOCK180 PAK ROCK Modulation of receptors’ output LIMK Myosin-II Colin Forward and reverse signaling Actin Trafcking and endocytosis NEURONAL GROWTH CONE Microtubules SnapShot: Axon Guidance R. -

Regulation of Cdc42 and Its Effectors in Epithelial Morphogenesis Franck Pichaud1,2,*, Rhian F
© 2019. Published by The Company of Biologists Ltd | Journal of Cell Science (2019) 132, jcs217869. doi:10.1242/jcs.217869 REVIEW SUBJECT COLLECTION: ADHESION Regulation of Cdc42 and its effectors in epithelial morphogenesis Franck Pichaud1,2,*, Rhian F. Walther1 and Francisca Nunes de Almeida1 ABSTRACT An overview of Cdc42 Cdc42 – a member of the small Rho GTPase family – regulates cell Cdc42 was discovered in yeast and belongs to a large family of small – polarity across organisms from yeast to humans. It is an essential (20 30 kDa) GTP-binding proteins (Adams et al., 1990; Johnson regulator of polarized morphogenesis in epithelial cells, through and Pringle, 1990). It is part of the Ras-homologous Rho subfamily coordination of apical membrane morphogenesis, lumen formation and of GTPases, of which there are 20 members in humans, including junction maturation. In parallel, work in yeast and Caenorhabditis elegans the RhoA and Rac GTPases, (Hall, 2012). Rho, Rac and Cdc42 has provided important clues as to how this molecular switch can homologues are found in all eukaryotes, except for plants, which do generate and regulate polarity through localized activation or inhibition, not have a clear homologue for Cdc42. Together, the function of and cytoskeleton regulation. Recent studies have revealed how Rho GTPases influences most, if not all, cellular processes. important and complex these regulations can be during epithelial In the early 1990s, seminal work from Alan Hall and his morphogenesis. This complexity is mirrored by the fact that Cdc42 can collaborators identified Rho, Rac and Cdc42 as main regulators of exert its function through many effector proteins. -

Two Locus Inheritance of Non-Syndromic Midline Craniosynostosis Via Rare SMAD6 and 4 Common BMP2 Alleles 5 6 Andrew T
1 2 3 Two locus inheritance of non-syndromic midline craniosynostosis via rare SMAD6 and 4 common BMP2 alleles 5 6 Andrew T. Timberlake1-3, Jungmin Choi1,2, Samir Zaidi1,2, Qiongshi Lu4, Carol Nelson- 7 Williams1,2, Eric D. Brooks3, Kaya Bilguvar1,5, Irina Tikhonova5, Shrikant Mane1,5, Jenny F. 8 Yang3, Rajendra Sawh-Martinez3, Sarah Persing3, Elizabeth G. Zellner3, Erin Loring1,2,5, Carolyn 9 Chuang3, Amy Galm6, Peter W. Hashim3, Derek M. Steinbacher3, Michael L. DiLuna7, Charles 10 C. Duncan7, Kevin A. Pelphrey8, Hongyu Zhao4, John A. Persing3, Richard P. Lifton1,2,5,9 11 12 1Department of Genetics, Yale University School of Medicine, New Haven, CT, USA 13 2Howard Hughes Medical Institute, Yale University School of Medicine, New Haven, CT, USA 14 3Section of Plastic and Reconstructive Surgery, Department of Surgery, Yale University School of Medicine, New Haven, CT, USA 15 4Department of Biostatistics, Yale University School of Medicine, New Haven, CT, USA 16 5Yale Center for Genome Analysis, New Haven, CT, USA 17 6Craniosynostosis and Positional Plagiocephaly Support, New York, NY, USA 18 7Department of Neurosurgery, Yale University School of Medicine, New Haven, CT, USA 19 8Child Study Center, Yale University School of Medicine, New Haven, CT, USA 20 9The Rockefeller University, New York, NY, USA 21 22 ABSTRACT 23 Premature fusion of the cranial sutures (craniosynostosis), affecting 1 in 2,000 24 newborns, is treated surgically in infancy to prevent adverse neurologic outcomes. To 25 identify mutations contributing to common non-syndromic midline (sagittal and metopic) 26 craniosynostosis, we performed exome sequencing of 132 parent-offspring trios and 59 27 additional probands. -

Regulation of the Mammalian Target of Rapamycin Complex 2 (Mtorc2)
Regulation of the Mammalian Target Of Rapamycin Complex 2 (mTORC2) Inauguraldissertation Zur Erlangung der Würde eines Doktors der Philosophie vorgelegt der Philosophisch-Naturwissenschaftlichen Fakultät der Universität Basel von Klaus-Dieter Molle aus Heilbronn, Deutschland Basel, 2006 Genehmigt von der Philosophisch-Naturwissenschaftlichen Fakultät Auf Antrag von Prof. Michael N. Hall und Prof. Markus Affolter. Basel, den 21.11.2006 Prof. Hans-Peter Hauri Dekan Summary The growth controlling mammalian Target of Rapamycin (mTOR) is a conserved Ser/Thr kinase found in two structurally and functionally distinct complexes, mTORC1 and mTORC2. The tumor suppressor TSC1-TSC2 complex inhibits mTORC1 by acting on the small GTPase Rheb, but the role of TSC1-TSC2 and Rheb in the regulation of mTORC2 is unclear. Here we examined the role of TSC1-TSC2 in the regulation of mTORC2 in human embryonic kidney 293 cells. Induced knockdown of TSC1 and TSC2 (TSC1/2) stimulated mTORC2-dependent actin cytoskeleton organization and Paxillin phosphorylation. Furthermore, TSC1/2 siRNA increased mTORC2-dependent Ser473 phosphorylation of plasma membrane bound, myristoylated Akt/PKB. This suggests that loss of Akt/PKB Ser473 phosphorylation in TSC mutant cells, as reported previously, is due to inhibition of Akt/PKB localization rather than inhibition of mTORC2 activity. Amino acids and overexpression of Rheb failed to stimulate mTORC2 signaling. Thus, TSC1-TSC2 also inhibits mTORC2, but possibly independently of Rheb. Our results suggest that mTORC2 hyperactivation may contribute to the pathophysiology of diseases such as cancer and Tuberous Sclerosis Complex. i Acknowledgement During my PhD studies in the Biozentrum I received a lot of support from many people around me who I mention here to express my gratefulness. -

The Atypical Guanine-Nucleotide Exchange Factor, Dock7, Negatively Regulates Schwann Cell Differentiation and Myelination
The Journal of Neuroscience, August 31, 2011 • 31(35):12579–12592 • 12579 Cellular/Molecular The Atypical Guanine-Nucleotide Exchange Factor, Dock7, Negatively Regulates Schwann Cell Differentiation and Myelination Junji Yamauchi,1,3,5 Yuki Miyamoto,1 Hajime Hamasaki,1,3 Atsushi Sanbe,1 Shinji Kusakawa,1 Akane Nakamura,2 Hideki Tsumura,2 Masahiro Maeda,4 Noriko Nemoto,6 Katsumasa Kawahara,5 Tomohiro Torii,1 and Akito Tanoue1 1Department of Pharmacology and 2Laboratory Animal Resource Facility, National Research Institute for Child Health and Development, Setagaya, Tokyo 157-8535, Japan, 3Department of Biological Sciences, Tokyo Institute of Technology, Midori, Yokohama 226-8501, Japan, 4IBL, Ltd., Fujioka, Gumma 375-0005, Japan, and 5Department of Physiology and 6Bioimaging Research Center, Kitasato University School of Medicine, Sagamihara, Kanagawa 252-0374, Japan In development of the peripheral nervous system, Schwann cells proliferate, migrate, and ultimately differentiate to form myelin sheath. In all of the myelination stages, Schwann cells continuously undergo morphological changes; however, little is known about their underlying molecular mechanisms. We previously cloned the dock7 gene encoding the atypical Rho family guanine-nucleotide exchange factor (GEF) and reported the positive role of Dock7, the target Rho GTPases Rac/Cdc42, and the downstream c-Jun N-terminal kinase in Schwann cell migration (Yamauchi et al., 2008). We investigated the role of Dock7 in Schwann cell differentiation and myelination. Knockdown of Dock7 by the specific small interfering (si)RNA in primary Schwann cells promotes dibutyryl cAMP-induced morpholog- ical differentiation, indicating the negative role of Dock7 in Schwann cell differentiation. It also results in a shorter duration of activation of Rac/Cdc42 and JNK, which is the negative regulator of myelination, and the earlier activation of Rho and Rho-kinase, which is the positive regulator of myelination. -

Related Malignant Phenotypes in the Nf1-Deficient MPNST
Published OnlineFirst February 19, 2013; DOI: 10.1158/1541-7786.MCR-12-0593 Molecular Cancer Genomics Research RAS/MEK–Independent Gene Expression Reveals BMP2- Related Malignant Phenotypes in the Nf1-Deficient MPNST Daochun Sun1, Ramsi Haddad2,3, Janice M. Kraniak2, Steven D. Horne1, and Michael A. Tainsky1,2 Abstract Malignant peripheral nerve sheath tumor (MPNST) is a type of soft tissue sarcoma that occurs in carriers of germline mutations in Nf1 gene as well as sporadically. Neurofibromin, encoded by the Nf1 gene, functions as a GTPase-activating protein (GAP) whose mutation leads to activation of wt-RAS and mitogen-activated protein kinase (MAPK) signaling in neurofibromatosis type I (NF1) patients' tumors. However, therapeutic targeting of RAS and MAPK have had limited success in this disease. In this study, we modulated NRAS, mitogen-activated protein/extracellular signal–regulated kinase (MEK)1/2, and neurofibromin levels in MPNST cells and determined gene expression changes to evaluate the regulation of signaling pathways in MPNST cells. Gene expression changes due to neurofibromin modulation but independent of NRAS and MEK1/2 regulation in MPNST cells indicated bone morphogenetic protein 2 (Bmp2) signaling as a key pathway. The BMP2-SMAD1/5/8 pathway was activated in NF1-associated MPNST cells and inhibition of BMP2 signaling by LDN-193189 or short hairpin RNA (shRNA) to BMP2 decreased the motility and invasion of NF1-associated MPNST cells. The pathway-specific gene changes provide a greater understanding of the complex role of neurofibromin in MPNST pathology and novel targets for drug discovery. Mol Cancer Res; 11(6); 616–27. -

A Rac/Cdc42 Exchange Factor Complex Promotes Formation of Lateral filopodia and Blood Vessel Lumen Morphogenesis
ARTICLE Received 1 Oct 2014 | Accepted 26 Apr 2015 | Published 1 Jul 2015 DOI: 10.1038/ncomms8286 OPEN A Rac/Cdc42 exchange factor complex promotes formation of lateral filopodia and blood vessel lumen morphogenesis Sabu Abraham1,w,*, Margherita Scarcia2,w,*, Richard D. Bagshaw3,w,*, Kathryn McMahon2,w, Gary Grant2, Tracey Harvey2,w, Maggie Yeo1, Filomena O.G. Esteves2, Helene H. Thygesen2,w, Pamela F. Jones4, Valerie Speirs2, Andrew M. Hanby2, Peter J. Selby2, Mihaela Lorger2, T. Neil Dear4,w, Tony Pawson3,z, Christopher J. Marshall1 & Georgia Mavria2 During angiogenesis, Rho-GTPases influence endothelial cell migration and cell–cell adhesion; however it is not known whether they control formation of vessel lumens, which are essential for blood flow. Here, using an organotypic system that recapitulates distinct stages of VEGF-dependent angiogenesis, we show that lumen formation requires early cytoskeletal remodelling and lateral cell–cell contacts, mediated through the RAC1 guanine nucleotide exchange factor (GEF) DOCK4 (dedicator of cytokinesis 4). DOCK4 signalling is necessary for lateral filopodial protrusions and tubule remodelling prior to lumen formation, whereas proximal, tip filopodia persist in the absence of DOCK4. VEGF-dependent Rac activation via DOCK4 is necessary for CDC42 activation to signal filopodia formation and depends on the activation of RHOG through the RHOG GEF, SGEF. VEGF promotes interaction of DOCK4 with the CDC42 GEF DOCK9. These studies identify a novel Rho-family GTPase activation cascade for the formation of endothelial cell filopodial protrusions necessary for tubule remodelling, thereby influencing subsequent stages of lumen morphogenesis. 1 Institute of Cancer Research, Division of Cancer Biology, 237 Fulham Road, London SW3 6JB, UK. -

NICU Gene List Generator.Xlsx
Neonatal Crisis Sequencing Panel Gene List Genes: A2ML1 - B3GLCT A2ML1 ADAMTS9 ALG1 ARHGEF15 AAAS ADAMTSL2 ALG11 ARHGEF9 AARS1 ADAR ALG12 ARID1A AARS2 ADARB1 ALG13 ARID1B ABAT ADCY6 ALG14 ARID2 ABCA12 ADD3 ALG2 ARL13B ABCA3 ADGRG1 ALG3 ARL6 ABCA4 ADGRV1 ALG6 ARMC9 ABCB11 ADK ALG8 ARPC1B ABCB4 ADNP ALG9 ARSA ABCC6 ADPRS ALK ARSL ABCC8 ADSL ALMS1 ARX ABCC9 AEBP1 ALOX12B ASAH1 ABCD1 AFF3 ALOXE3 ASCC1 ABCD3 AFF4 ALPK3 ASH1L ABCD4 AFG3L2 ALPL ASL ABHD5 AGA ALS2 ASNS ACAD8 AGK ALX3 ASPA ACAD9 AGL ALX4 ASPM ACADM AGPS AMELX ASS1 ACADS AGRN AMER1 ASXL1 ACADSB AGT AMH ASXL3 ACADVL AGTPBP1 AMHR2 ATAD1 ACAN AGTR1 AMN ATL1 ACAT1 AGXT AMPD2 ATM ACE AHCY AMT ATP1A1 ACO2 AHDC1 ANK1 ATP1A2 ACOX1 AHI1 ANK2 ATP1A3 ACP5 AIFM1 ANKH ATP2A1 ACSF3 AIMP1 ANKLE2 ATP5F1A ACTA1 AIMP2 ANKRD11 ATP5F1D ACTA2 AIRE ANKRD26 ATP5F1E ACTB AKAP9 ANTXR2 ATP6V0A2 ACTC1 AKR1D1 AP1S2 ATP6V1B1 ACTG1 AKT2 AP2S1 ATP7A ACTG2 AKT3 AP3B1 ATP8A2 ACTL6B ALAS2 AP3B2 ATP8B1 ACTN1 ALB AP4B1 ATPAF2 ACTN2 ALDH18A1 AP4M1 ATR ACTN4 ALDH1A3 AP4S1 ATRX ACVR1 ALDH3A2 APC AUH ACVRL1 ALDH4A1 APTX AVPR2 ACY1 ALDH5A1 AR B3GALNT2 ADA ALDH6A1 ARFGEF2 B3GALT6 ADAMTS13 ALDH7A1 ARG1 B3GAT3 ADAMTS2 ALDOB ARHGAP31 B3GLCT Updated: 03/15/2021; v.3.6 1 Neonatal Crisis Sequencing Panel Gene List Genes: B4GALT1 - COL11A2 B4GALT1 C1QBP CD3G CHKB B4GALT7 C3 CD40LG CHMP1A B4GAT1 CA2 CD59 CHRNA1 B9D1 CA5A CD70 CHRNB1 B9D2 CACNA1A CD96 CHRND BAAT CACNA1C CDAN1 CHRNE BBIP1 CACNA1D CDC42 CHRNG BBS1 CACNA1E CDH1 CHST14 BBS10 CACNA1F CDH2 CHST3 BBS12 CACNA1G CDK10 CHUK BBS2 CACNA2D2 CDK13 CILK1 BBS4 CACNB2 CDK5RAP2 -

Targeting PH Domain Proteins for Cancer Therapy
The Texas Medical Center Library DigitalCommons@TMC The University of Texas MD Anderson Cancer Center UTHealth Graduate School of The University of Texas MD Anderson Cancer Biomedical Sciences Dissertations and Theses Center UTHealth Graduate School of (Open Access) Biomedical Sciences 12-2018 Targeting PH domain proteins for cancer therapy Zhi Tan Follow this and additional works at: https://digitalcommons.library.tmc.edu/utgsbs_dissertations Part of the Bioinformatics Commons, Medicinal Chemistry and Pharmaceutics Commons, Neoplasms Commons, and the Pharmacology Commons Recommended Citation Tan, Zhi, "Targeting PH domain proteins for cancer therapy" (2018). The University of Texas MD Anderson Cancer Center UTHealth Graduate School of Biomedical Sciences Dissertations and Theses (Open Access). 910. https://digitalcommons.library.tmc.edu/utgsbs_dissertations/910 This Dissertation (PhD) is brought to you for free and open access by the The University of Texas MD Anderson Cancer Center UTHealth Graduate School of Biomedical Sciences at DigitalCommons@TMC. It has been accepted for inclusion in The University of Texas MD Anderson Cancer Center UTHealth Graduate School of Biomedical Sciences Dissertations and Theses (Open Access) by an authorized administrator of DigitalCommons@TMC. For more information, please contact [email protected]. TARGETING PH DOMAIN PROTEINS FOR CANCER THERAPY by Zhi Tan Approval page APPROVED: _____________________________________________ Advisory Professor, Shuxing Zhang, Ph.D. _____________________________________________